Е74 код по мкб

Связанные заболевания и их лечение
Описания заболеваний
Национальные рекомендации по лечению
Стандарты мед. помощи
Содержание
- Синонимы диагноза
- Описание
- Дополнительные факты
- Причины
- Классификация
- Симптомы
- Диагностика
- Лечение
- Прогноз
- Основные медицинские услуги
- Клиники для лечения
Названия
E74,0 Болезни накопления гликогена.
E74.0 Болезни накопления гликогена
Синонимы диагноза
Болезни накопления гликогена, гликогенозы, помпе болезнь.
Описание
Болезнь Помпе. Редкая наследственная патология, одна из форм лизосомных болезней накопления, характеризующаяся нарушением процессов расщепления гликогена в нервных и мышечных клетках (скелетные мышцы, миокард). Симптомы заболевания довольно вариабельны по времени своего проявления и выраженности у разных больных, традиционно наблюдается прогрессирующая мышечная слабость, при некоторых формах – кардиомегалия с дилятационной кардиомиопатией. Диагностика болезни Помпе производится на основании данных наследственного анамнеза, гистологического и гистохимического изучения мышечных тканей, биохимического анализа крови, а также генетических исследований. Лечение в настоящий момент может производиться с помощью фермент-заместительной терапии, однако эффективность этой методики неодинакова у разных пациентов.
E74.0 Болезни накопления гликогена
Дополнительные факты
Болезнь Помпе (гликогеноз 2-го типа, недостаточность кислой альфа-глюкозидазы) – наследственное заболевание, при котором из-за нарушения процессов обмена гликогена происходит повреждение нервных и мышечных тканей. Впервые было описано в 1932 году голландским ученым И. Помпе, с тех пор официально зарегистрировано более 50 случаев патологии. Болезнь Помпе с равной степенью вероятности поражает как мужчин, так и женщин, встречаемость колеблется от 1:60000 (взрослая форма) до 1:140000 (ранняя, или инфантильная форма). Является одной из немногих лизосомных болезней накопления, в отношении которой было разработано эффективное специфическое лечение, одобренное в США в 2006 году и в России – в 2013 г. Однако стоимость этиотропной терапии болезни Помпе крайне высока и составляет несколько сотен тысяч долларов в год. Смертность в случае отсутствия лечения зависит от формы патологии – детская инфантильная форма часто приводит к летальному исходу на 1-2-м году жизни ребенка, при типе болезни с отсроченным началом нарастание симптомов идет намного медленнее.
Причины
Болезнь Помпе является классическим гликогенозом, при ней в тканях скелетных мышц, миокарда и отчасти нервной системы формируются отложения гликогена по причине невозможности его расщепления. Это происходит в результате мутации гена GAA, расположенного на 17-й хромосоме – он кодирует последовательность кислой альфа-1,4-глюкозидазы или мальтазы. Это один из ключевых ферментов лизосом, участвующий в расщеплении молекулы гликогена на более простые отрезки, которые, в конечном итоге, деградируют до глюкозы, вступающей в энергетический обмен клетки. Так как гликоген является важным депо энергии для таких структур, как скелетные мышцы, миокард, печень и нервная ткань, проявления болезни Помпе сводятся именно к патологическим изменениям данных органов.
В результате подобных изменений сначала возникает дефицит энергии в клетках – потребности тканей в глюкозе покрываются только за счет ее поступления из крови. Кроме того, в лизосомах при болезни Помпе начинает накапливаться гликоген, формируя крупные включения в виде вакуолей, в дальнейшем приводя к дистрофии и повреждению клеток. Наследование дефектных вариантов гена GAA происходит по аутосомно-рецессивному типу. Наличие нескольких форм заболевания предположительно объясняется разными типами мутаций вышеуказанного гена. Возможно, при некоторых дефектах происходит не полное исчезновение, а лишь снижение активности кислой альфа-1,4-глюкозидазы, что и приводит к более позднему развитию болезни Помпе и медленному прогрессированию заболевания. Определение формы патологии играет важную роль для составления ее прогноза и схемы лечения.
Классификация
На сегодняшний день специалисты выделяют несколько основных форм болезни Помпе, основное различие между которыми заключается в сроках начала заболевания и выраженности симптомов. В большинстве случаев, с гликогенозом 2-го типа сталкиваются врачи-педиатры, однако имеется тип заболевания, выявляемый у взрослых.
• Ранняя инфантильная форма. Такая разновидность болезни Помпе считается наиболее тяжелой. Выявляется еще на первых месяцах жизни, симптомы миопатии, поражения печени (гепатомегалия) и сердца (кардиомиопатия) достаточно быстро прогрессируют. Обычно больные с ранней инфантильной формой болезни Помпе умирают от сердечной или дыхательной недостаточности в возрасте до года.
• Поздняя инфантильная форма. Первые симптомы возникают в возрасте 1 — 3 года, скорость их прогрессирования также намного медленнее. При данном типе болезни Помпе наиболее выражены поражения миокарда, смерть наступает к подростковому возрасту от сердечной недостаточности.
• Ювенильная форма болезни Помпе развивается в возрасте 6-10 лет. Так же, как и в предыдущем варианте, основным органом-мишенью болезни становится сердце, смерть от нарастающей сердечной недостаточности наступает к 20 годам.
• Взрослая форма болезни Помпе. Манифестация симптомов происходит в 20 — 40 лет. Ведущим симптомом является медленно прогрессирующая миопатия, поражения печени практически никогда не регистрируются, в некоторых случаях возможны незначительные нарушения миокарда. При этом типе болезни Помпе больные во многих случаях доживают до старости, лишь иногда возможен более ранний летальный исход из-за дыхательной или сердечной недостаточности.
Методами современной генетики на сегодняшний момент не определена взаимосвязь между отдельными типами мутаций гена GAA и формами болезни Помпе. Возможно, причина такой вариабельности проявлений лежит совсем в другом – в литературе описаны семейные случаи заболевания, когда у родственников регистрировались различные формы патологии. Изучение закономерностей, приводящих к развитию болезни Помпе определенного типа, сильно осложняется относительной редкостью данного синдрома.
Симптомы
Проявления болезни Помпе довольно сильно отличаются при различных формах заболевания. Ранний инфантильный тип характеризуется выраженной мышечной слабостью младенца, снижением его двигательной активности, плаксивостью. В педиатрии при осмотре такого больного часто выявляется задержка психомоторного развития, различная степень увеличения печени, пальпация иногда выявляет гипертрофию мышц, которые, однако, при этом довольно слабые. При дальнейшем развитии болезни Помпе возникают проблемы с кормлением из-за слабости сосательной мускулатуры, выявляется дисфагия и, как итог всего этого – гипотрофия. Нарастающая кардиомиопатия и слабость дыхательной мускулатуры со временем приводят к смерти ребенка.
Поздняя инфантильная и ювенильная формы болезни Помпе протекают практически одинаково, различается только срок появления симптомов патологии. Как правило, выявляется мышечная слабость, признаки кардиомиопатии. Со временем начинает формироваться выраженная дистрофия скелетной мускулатуры, кардиомегалия, на этом фоне начинают увеличиваться печень и селезенка. Длительность течения этих форм болезни Помпе составляет около 10-12 лет, после чего, при отсутствии лечения, наступает летальный исход из-за декомпенсированной сердечной недостаточности. Косвенным симптомом будет являться большая частота простудных заболеваний с легочными осложнениями, ночное апноэ, головные боли по утрам.
Диагностика
Выявление болезни Помпе можно производить многочисленными клиническими методиками – биохимическим анализом крови, изучением биоптата мышц, культур фибробластов или лейкоцитов больного, традиционными исследованиями (осмотр, ЭКГ, ЭхоКГ). При осмотре часто обнаруживается слабость и дистрофия мышц, при вовлечении в патологический процесс внутренних органов – гепато- и спленомегалия. На электрокардиограмме регистрируется укорочение интервала PQ, расширение комплекса QRS, обусловленного увеличением размеров миокарда. По этой же причине увеличивается длительность фазы реполяризации желудочков, что проявляется инверсией зубца T. Эхографическое исследование сердца показывает резкое увеличение его размеров за счет значительного утолщения стенок желудочков. При взрослой форме болезни Помпе вышеуказанные изменения миокарда могут не выявляться.
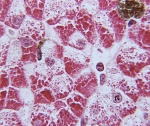
Биохимический анализ крови позволяет обнаружить как специфические, так и косвенные признаки заболевания. Специфическим исследованием будет определение активности кислой альфа-1,4-глюкозидазы в плазме крови, которая при болезни Помпе будет резко снижена. Косвенным указанием на наличие патологии является резкое повышение активности креатинфосфокиназы, обусловленное поражением мышечной ткани. Гистологическое исследование биоптата мышц выявляет в миоцитах многочисленные включения гликогена, часто придающие им вид «пенистых клеток». Гистохимическое изучение при болезни Помпе выявляет резкое снижение активности кислой альфа-1,4-глюкозидазы в мышечной ткани, фибробластах и лейкоцитах.
Врачом-генетиком может быть проведено генетическое определение болезни Помпе – оно производится методом прямого секвенирования последовательности гена GAA с целью выявления дефектных участков. Кроме того, может помочь в диагностике заболевания и составление наследственного анамнеза. Генетическая диагностика болезни Помпе включает в себя секвенирование GAA и у фенотипически здоровых родственников больного с целью выявления носительства патологического гена.
Лечение
На сегодняшний день единственным методом специфического лечения болезни Помпе является фермент-заместительная терапия с целью восполнения дефицита кислой альфа-1,4-глюкозидазы. Для этого используют препарат альфа алглюкозидазы производства США. Стоимость этого лечения крайне высока (годовой курс стоит 100-400 тысяч долларов), однако его эффективность неодинакова у разных больных. Других способов лечения болезни Помпе в настоящее время не существует.
Прогноз
Без лечения при инфантильных и ювенильной формах заболевания прогноз неблагоприятный, у взрослого типа – неопределенный. Профилактика возможна только путем своевременного выявления носительства болезни Помпе (в случае наличия патологии у кровных родственников) и последующей генетической пренатальной диагностики.
Основные медуслуги по стандартам лечения | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Клиники для лечения с лучшими ценами
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Источник
Рубрика МКБ-10: E74.8
МКБ-10 / E00-E90 КЛАСС IV Болезни эндокринной системы, расстройства питания и нарушения обмена веществ / E70-E90 Нарушения обмена веществ / E74 Другие нарушения обмена углеводов
Определение и общие сведения[править]
Семейная почечная глюкозурия
Семейная почечная глюкозурия характеризуется наличием стойкой изолированной глюкозурии в отсутствие как генерализованной дисфункции проксимальных почечных канальцев, так и гипергликемии. Семейная почечная глюкозурия обычно считается доброкачественным заболеванием, поскольку большинство пациентов не имеют тяжелые клинические последствия.
Этиология и патогенез[править]
Семейная почечная глюкозурия вызвана мутациями с потерей функции в гене SLC5A2 (16p11.2).
Увеличение экскреции глюкозы с мочой обусловлено разнообразными причинами. У здоровых лиц глюкозурия не выражена, её невозможно определить рутинными лабораторными методами, а увеличение выраженности глюкозурии, например, при проведении теста толерантности к глюкозе носит транзиторный характер.
Почечная глюкозурия часто является самостоятельным заболеванием; её, как правило, обнаруживают случайно; полиурию и полидипсию наблюдают крайне редко. Иногда почечной глюкозурии сопутствуют другие тубулопатии, в том числе в составе синдрома Фанкони.
Среди возможных причин почечных глюкозурий типов 1 и 2 обсуждают мутации одного из канальцевых белков-переносчиков, реабсорбирующих глюкозу вместе с двумя ионами натрия. Однако разграничение этих вариантов на генетическом уровне затруднительно, поскольку в одной семье диагностируют случаи почечной глюкозурии как 1-го, так и 2-го типа.
Патогенез
1. При почечной глюкозурии типа 1 наблюдают значительное снижение реабсорбции глюкозы в проксимальных канальцах при относительно сохранных величинах клубочковой фильтрации. Коэффициент отношения максимальной реабсорбции глюкозы к СКФ у пациентов с почечной глюкозурией типа 1 снижен.
2. Почечная глюкозурия типа 2 характеризуется значительным увеличением порога реабсорбции глюкозы эпителиоцитами проксимальных канальцев. Коэффициент отношения максимальной реабсорбции глюкозы к СКФ (скорость клубочковой фильтрации ) близок к нормальному.
3. Крайне редко наблюдают почечную глюкозурию типа 0, при которой способность эпителиоцитов проксимальных канальцев реабсорбировать глюкозу полностью отсутствует. Развитие глюкозурии связывают с мутацией, обусловливающей отсутствие или значительный дефект, сопровождающийся полной утратой реабсорбирующей функции, канальцевых белков, транспортирующих глюкозу. У этих больных величины глюкозурии достигают особенно высоких цифр.
Существуют и более редкие варианты почечной глюкозурии. Описано сочетание почечной глюкозурии типа 1 с глицинурией и гиперфосфатурией; при этом другие признаки синдрома Фанкони, в том числе аминоацидурия, отсутствуют.
При комбинации почечной глюкозурии с глицинурией больные нередко страдают муковисцидозом. Считают, что этот вариант тубулопатии наследуют по аутосомно-доминантному типу.
Идентифицирована мутация, обусловливающая значительное снижение активности кишечного траспортёра для глюкозы и галактозы. Одновременно у этих больных обнаруживают нарушения реабсорбции глюкозы в канальцах, чаще сходные с почечной глюкозурией типа 2.
Почечную глюкозурию наблюдают у беременных. Развитие её обусловлено значительным физиологическим увеличением СКФ при относительно стабильных показателях максимальной реабсорбции глюкозы. Глюкозурия беременных носит транзиторный характер.
Клинические проявления[править]
Другие уточненные нарушения обмена углеводов: Диагностика[править]
Лабораторные исследования
Почечную глюкозурию диагностируют по наличию глюкозы в моче натощак при нормальном уровне гликемии. Почечное происхождение глюко-зурии подтверждают обнаружением глюкозы не менее чем в трёх порциях мочи и отсутствием изменений в гликемической кривой при проведении теста толерантности к глюкозе.
При почечной глюкозурии величина экскреции глюкозы с мочой варьирует от 500 мг/сут до 100 г/сут и более, у большинства пациентов она составляет 1-30 г/сут.
Дифференциальный диагноз[править]
У беременных необходимо проводить дифференциальную диагностику глюкозурии с сахарным диабетом беременных.
Другие уточненные нарушения обмена углеводов: Лечение[править]
Лечение почечной глюкозурии предусматривает подбор сбалансированного рациона, содержащего достаточное количество углеводов. При полиурии для предупреждения потери калия рекомендуют употребление сухофруктов.
Профилактика[править]
Прочее[править]
Оксалурия
Первичная гипероксалурия является редким расстройством метаболизма гликоксилата, характеризующееся накоплением оксалатов. Гипероксалурия может проявляться единичными почечными камнями, рецидивирующим нефролитиазом и нефрокальцинозом вплоть до терминальной стадии хронической почечной недостаточности и системного оксалоза.
Манифестация гипероксалурии может наблюдаться в любом возрасте. Хорошо известны три варианта патологии: первичная гипероксалурия типов 1, 2 и 3.
Наиболее частая форма тип 1 из-за дефицита глиоксилат аминотрансферазы — пероксисомального фермента L-аланина печени, вызванного мутациями в гене AGXT (2q37,3).
Оксалурия — стойкое выделение с мочой кристаллов оксалата кальция. Заболевание связано с нарушением выделения почками защитных коллоидов, поддерживающих в норме щавелевую кислоту в растворённом состоянии. Оксалаты кальция выпадают при любом рН мочи, чаще при 5,4-6,6.
При построении диетического рациона для больных оксалурией следует учитывать, что введение с пищей продуктов, богатых щавелевой кислотой, повышает выделение оксалатов с мочой. Из рациона исключают продукты с высоким содержанием щавелевой кислоты и её солей: щавель, шпинат, свёклу, картофель, бобы, ревень, инжир, петрушку, некоторые ягоды (сливу, землянику, крыжовник), чай, какао, кофе и др.
Выведению оксалатов из организма способствуют яблоки, груши, айва, листья грушевого дерева, винограда, чёрной смородины (в виде отвара). Отвары, приготовленные из кожуры фруктов, усиливают выведение щавелевой кислоты из организма.
Диетический рацион больного оксалурией включает следующие продукты: белый и чёрный хлеб, масло животное и растительное, молоко, творог, сметану, яйца, сыр, вегетарианские супы (из разрешаемых овощей и фруктов), молочные супы, мясо, рыбу и птицу в отварном виде в ограниченном количестве (по 150 г через день), блюда из круп и теста.
Больному оксалурией разрешается употребление достаточного количества жидкости (до 2 л) и соков из свежих овощей и фруктов. Несколько ограничивают в рационе поваренную соль и углеводы. Больным оксалурией можно рекомендовать диету № 5 с ограничением углеводов до 300 г. Из овощей и фруктов рекомендуют цветную и белокочанную капусту, чечевицу, горох, зелёный горошек, репу, спаржу, огурцы, яблоки, груши, абрикосы, персики, виноград, кизил, айву. При обострении заболевания необходимо ограничить в рационе молоко и молочные продукты, содержащие много кальция.
Пентозурия
Синонимы: дефицит ксилитолдегидрогеназы
Определение и общие сведения
Пентозурия — врожденное нарушение обмена веществ, которое характеризуется экскрецией с мочой от 1 до 4 г L-ксилулозы в день. Передается аутосомно-рецессивно.
Пентозурия обнаруживается в основном в популяции евреев-ашкенази с предполагаемой частотой гетерозиготной мутации 1/79.
Этиология и патогенез
Пентозурия вызывается мутациями в гене DCXR на хромосоме 17, кодирующий фермент L-ксилоз редуктазу (или L-ксилитолдегидрогеназа), который катализирует превращение 1-ксилулозы в ксилит.
Клинические проявления
Пентозурия является доброкачественным состоянием и не проявляет себя клинически. Единственной биологической особенностью является постоянное выделение L-ксилулозы с мочой, которые могут быть ошибочно приняты за проявления глюкозурии.
Изолированная недостаточность глицеринкиназы
Синонимы: гиперглицеринемия
Изолированная недостаточность глицеринкиназы является очень редким Х-сцепленным нарушением метаболизма глицерина, которое биохимически характеризуется повышенным уровнем глицерина в плазме и моче, а клинически — различными нейрометаболическими проявлениями в зависимости от возраста манифестации заболевания. Тяжесть гиперглицеринемии варьирует от жизнеугрожающего метаболического криза у детей до бессимптомной формы у взрослых. Выделяют инфантильную, ювенильную и взрослую формы недостаточности глицеринкиназы.
Дефицит трансальдолазы
Дефицит трансальдолазы является врожденным нарушением метаболизма пентозофосфата, который проявляется в неонатальном или антенатальном периоде с водянкой плода, гепатоспленомегалией, печеночной дисфункцией, тромбоцитопенией, анемией и почечными и сердечными аномалиями.
Описано менее 10 случаев дефицита трансальдолазы у детей, родившимися в близкородственных браках у родителей турецкого и арабского происхождения.
Дефицит трансальдолазы вызван мутациями в гене трансальдолазы TALDO1 (11p15.5-p15.4).
Источники (ссылки)[править]
Нефрология [Электронный ресурс] / Под ред. Е.М. Шилова. — 2-е изд., испр. и доп. — М. : ГЭОТАР-Медиа, 2010. — https://www.rosmedlib.ru/book/ISBN9785970416419.html
Клиническая диетология [Электронный ресурс] / В. П. Шевченко ; под ред. В. Т. Ивашкина. — М. : ГЭОТАР-Медиа, 2014. — (Серия «Библиотека врача-специалиста»). — https://www.rosmedlib.ru/book/ISBN9785970430088.html
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
Источник