Дисгенезия гонад код по мкб

Описание болезни
Дисгенезия гонад – неполное развитие половых желез, т.е. недоразвитие яичек или яичников.
Дисгенезия гонад относится к генетически обусловленным формам задержки полового развития (ЗПР) человека.
1. Типичная дисгенезия гонад (синдром Шерешевского-Тернера, Морганьи-Тернера-Олбрайта, Улльрихи-Бонневи, яичниковый карликовый рост, агенезия половых желез). Синдром впервые описан в 1925 году.
Клинические признаки: наличие лимфатического отека стоп, кистей, верхней части туловища, шеи у новорожденных; низкий рост; коренастость; широкая бочкообразная грудная клетка с широко расставленными, часто втянутыми сосками и сформированной грудиной; задержка появления вторичных половых признаков и менструаций, наружные половые органы гипопластичны, клитор нормальных, размеров, матка резко гипопластичная, влагалище длинное и узкое; шея короткая, низкая линия роста волос; лицо своеобразное – дети имеют вид стариков; недоразвитие нижней челюсти; аркообразное небо, зубы деформированы; пигментация век, птоз, косоглазие, эпикантус, поперечные складки на шее; аномалии костной системы (деформация позвоночника, остепороз), сердечно-сосудистой, выделительной систем; при кольпоцитологии атрофичный тип мазка, экскреция 17-КС в норме или уменьшена; половой хроматин отсутствует или процент резко снижен; при стертой форме имеется умеренно выраженное половое недоразвитие, первичная или вторичная аменорея; первичное бесплодие; низкий рост; вальгусная девиация локтевых суставов; короткая шея.
2. Чистая дисгенезия гонад (синдром Свайера). Синдром впервые описан в 1955 году.
Клинические признаки: наличие полового инфантилизма, высокого роста, соматических аномалий; женщины высокого роста, часто имеют евнухоидный тип тела; появление вторичных половых признаков не наблюдается; молоч¬ные железы отсутствуют или слабо развиты и пальпируются в виде комоч¬ков под ареолами; наружные и внутренние гениталии женского типа, гипоп-ластичны; оволосение скудное, характер его распределения женский; на месте яичников расположены гонадальные тяжи; у большинства кариотип неизменен, но встречается как мужской (46, ХУ), так и женский (46, XX); все больные являются лицами женского пола, который им присваивается с мо¬мента рождения, до периода полового созревания не подозревают о своем заболевании; у лиц с кариотнпом 46, ХУ может наблюдаться гипертрофия клитора, иногда значительное, матка в виде рудимента; у лиц с кариотипом 46, XX клиническое проявление полового инфантилизма может быть менее выражено, в отдельных случаях могут наблюдаться менструальноподобные выделения.
3. Смешанная, или атипичная, дисгенезия гонад.
Представляет собой переходную форму от тип
Дисгенезия гонад в МКБ классификации:
—
Q50-Q56 Врожденные аномалии [пороки] половых органов
- Q50 Врожденные аномалии [пороки развития] яичников, фаллопиевых труб и широких связок
Светлана:25.07.2014
Здравствуйте! Моему сыну (3 года 10 мес) весной поставили диагноз — паховая грыжа и назначили операцию на 30 июля. Весной припухлость в области паха появлялась довольно часто, особенно при плаче. Но в последнее время я ее вижу редко и если она появляется, то ее размер стал раза в 3 меньше. Вчера были на консультации у хирурга. Он диагноз не подтвердил. В начале июля делали УЗИ — заключение — паховая грыжа справа — диаметр внутреннего пахового кольца справа 2 мм, слева 1,8 мм в положении лежа, стоя паховые каналы сомкнуты. При натуживании справа в верхней трети пахового канала определяется грыжевое выпячивание 13,4 на 5,8 мм, содержит брюшную жидкость. Посоветуйте, что нам делать: ложиться на операцию или пока наблюдаться?
Добрый день! К сожалению паховая грыжа лечится только хирургическим путем и откладывать операцию опасно. Детки в этом возрасте очень подвижны и есть высокий риск ущемления паховой грыжи, что влечет за собой тяжелые последствия. Не волнуйтесь, вовремя проведенное хирургическое лечение переносится хорошо, главное делать операцию на фоне полного здоровья. Будьте здоровы!

К каким врачам обращаться, если возникает Дисгенезия гонад:
Источник
Содержание :
- Что из себя представляет синдром
- Причины заболевания
- Клиническая картина синдрома
Синдром Шерешевского-Тернера (код по мкб 10 –Q00-Q99/Q90-Q99/Q96) – это заболевание, которое возникает вследствие хромосомных сбоев. Синдром Шерешевского-Тернера обусловлен полной или частичной моносомией X хромосомы, то есть, хромосома при синдроме Шерешевского-Тернера отсутствует. Клиническая картина синдрома проявляется множеством симптомов: складки кожи в виде крыльев на шее, низкий рост, недостаточное функционирование половых желез, деформации скелета.
Диагностика. Диагностировать патологию можно еще внутриутробно. После рождения диагноз ставится на основе данных осмотра и дополнительных обследований, а именно определение кариотипа и полового хроматина. Жизнь при синдроме Шерешевского-Тернера тесно связана с постоянной медицинской помощью. Лечение синдрома Шерешевского-Тернера обязательно.
Люди с синдромом Шерешевского-Тернера нуждаются в заместительной гормонотерапии и лечении сопутствующих пороков. Довольно часто им также проводят операции для улучшения внешнего вида, избавления от эстетических недостатков.
Что из себя представляет синдром
Заболевание синдром Шерешевского-Тернера – это геномная патология, первичная гонадная дисгенезия, которая возникает вследствие сбоев в половых хромосомах. Синдром Шерешевского-Тернера частота: одна из 3000 беременностей заканчивается рождением ребенка с данным синдромом. На самом деле больных плодов намного больше, но чаще всего с таким диагнозом , как синдром Шерешевского-Тернера продолжительность жизни плода не велика, многие не доживают до больших сроков беременности. Организм избавляется от генетически неправильных детей путем самопроизвольного выкидыша еще в первом триместре.
Кариотип девочки с синдромом Шерешевского-Тернера
В медицинской литературе синдром можно встретить и под другими названиями – синдром Ульриха-Тернера, синдром Шерешевского, синдром Тернера, синдром Шерешевского-Тернера 45 Х0и тд. Он назван фамилиями людей, которые изучали заболевание.
Чаще всего встречается синдром Шерешевского-Тернера у девочек , которые могут жить с ней практически нормальной жизнью. Реже встречается синдром Шерешевского-Тернера у мальчиков. К сожалению, представители мужского пола, в отличие от девочек, не способны выжить при отсутствии X-хромосомы.
Причины заболевания
Синдром Шерешевского-Тернера причины и признаки. Синдром Шерешевского-Тернера вызван структурными или количественными изменениями половой X-хромосомы, такими влияниями, как мутация. Около 60% аномалий припадает на абсолютную моносомию, то есть полное отсутствие одной хромосомы. Еще в 20% случаев определяются перестройки в структуре. Генотип претерпевает значительные изменения.
Формы синдрома Шерешевского-Тернера. Кариотип больных с синдромом Шерешевского-Тернера:
- Частичная или полная потеря длинного или короткого плеча (делеция);
- Перенесение участка хромосомы (транслокация);
- Утрата одного плеча с последующим дублированием оставшегося (изохромосома);
- Кольцевая X-хромосома.
Оставшиеся 20% занимает синдром Шерешевского-Тернера, мозаичная форма – наличие в одном организме клеток, которые являются генетически разными. Причиной появления синдрома Шерешевского-Тернера у мужчин является транслокация и мозаицизм хромосом — мозаичный синдром Шерешевского-Тернера.
Причины возникновения синдрома Шерешевского-Тернера различны
Доказано, что возраст матери никоим образом не влияет на риск возникновения патологии. Настоящими причинами изменения количества, качества или структуры хромосомы являются нарушения расхождения во время мейоза, которые приводят к анеуплоидии. Также к развитию синдрома Шерешевского-Тернера может провести неправильное дробление зиготы – возникает хромосомный мозаицизм. В подавляющем большинстве случаев при X-моносомии определяют отсутствие X-хромосомы, которая должна была быть передана от отца.
Беременность: признаки синдрома Шерешевского-Тернера
У плода с синдромом Шерешевского-Тернера значительно нарушается формирование половых желез. Часто возникают различные пороки внутренних органов. Вынашивание больного плода практически всегда характеризуется сильным токсикозом, наличием угрозы самопроизвольного аборта и преждевременных родов.
Клиническая картина синдрома
Обычно дети с синдромом Шерешевского-Тернера, рождаются раньше срока. Но даже в случае беременности, доношенной до 40 недель, вес новорожденного редко достигает 3 кг, а рост более 50 см.
Симптомы синдрома Шерешевского-Тернера
Сразу же после родов, можно заметить у ребенка признаки, которые характерны для заболевания:
- Укороченная шея;
- Птеригиум-синдром – кожные складки в виде крыльев на боках шеи;
- Нарушение оттока лимфы;
- Отеки на стопах и кистях;
- Наличие врожденных пороков сердечно-сосудистой системы.
Далее на первый план выступают проблемы с кормлением – у детей нарушено сосание, они часто срыгивают «фонтаном», находится в моторном возбуждении. На первых годах жизни можно заметить отставание в развитие – ребенок поздно начинает сидеть, ходить, говорить. Также для синдрома Шерешевского-Тернера характерно частое, повторное возникновение среднего отита, которое впоследствии приводит к кондуктивной тугоухости.
На момент полового созревания люди с синдромом Шерешевского-Тернера выглядят следующим образом: рост людей редко когда превышает 150 см. Месячные при синдроме Шерешевского-Тернера отсутствуют.
Такая болезнь, синдром Шерешевского-Тернера, имеет характерные фенотипические проявления.
Кроме этого, они имеют характерный внешний вид:
- Лицо обретает определенное выражение – «лицо Сфинкса»;
- Шея укорочена, присутствует птеригиум-синдром;
- Граница роста волос занижена;
- Челюсть недоразвита – микрогнатия;
- Уши увеличены, часто бывает лопоухость;
- Грудная клетка слишком широкая.
Жизнь с синдромом Шерешевского-Тернера непроста, при данной патологии значительно поражается костная система организма. Часто возникает сколиоз, дисплазия суставов, особенно тазобедренных, девиация локтей. На черепе наблюдается микрогнатия, неправильный прикус и готическое небо. В связи с недостаточным количеством эстрогена, люди с таким синдромом подвержены раннему возникновению остеопороза. У них часто случаются переломы позвоночника, костей кисти и шейки бедра.
Продолжительность жизни у людей с синдромом Шерешевского-Тернера
Сколько живут люди с синдромом Шерешевского-Тернера зависит от степени тяжести заболевания и от осложнений систем органов, которые возникают вследствие данной патологии. Со стороны сердечно-сосудистой системы встречаются такие пороки, как коарктация и аневризма аорты, дефекты межжелудочковой и межпредсердной перегородки. С такими пороками прогноз для жизни неблагоприятный. В почках часто бывает раздвоение лоханок, стеноз артерий, подковообразная форма самой почки. Подобные патологии приводят к артериальной гипертензии. У больных синдромом Шерешевского-Тернера нередко развивается косоглазие, близорукость и опущение века. К тому же они могут также страдать и дальтонизмом.
Умственные способности, как правило, сохраняются на должном уровне, но в некоторых случаях может наблюдаться олигофрения. Людей, страдающих синдромом Шерешевского-Тернера, часто сопровождают соматические заболевания – алопеция, микседема, витилиго, дефицит ферментов в тонком кишечнике, ожирение. Практически во всех случаях диагностируется диабет первого или второго типа и ишемическое поражение сердца. Доказано, что у таких пациентов рак толстого кишечника возникает в несколько раз чаще, чем у здоровых людей.
Характеристика женщин, страдающих данным заболеванием
Практически у 100% больных девушек диагностируется первичный гипогонадизм. Внутренние половые органы недоразвиты – матка гипоплазирована, а вместо яичников с двух сторон находятся фиброзные тяжи. Женщины с синдромом Шерешевского-Тернера обычно полностью стерильны – в яичниках отсутствуют фолликулы с яйцеклетками. Вульва видоизменена: большие губы похожи на мошонку, а малые и клитор недоразвиты. Девственная плева может отсутствовать. Некоторые задаются вопросом как приходят месячные с синдромом Шерешевского-Тернера — в период полового созревания обнаруживается первичная аменорея. Грудные железы развиты недостаточно, наблюдается незначительное оволосение в участке лобка и подмышек. Беременность, которая возникла природным путем, возможно только в случае мозаичной формы поражения X-хромосомы.
Мальчики при синдроме Шерешевского Тернера
Половые органы мужчины также недоразвиты. Яички гипоплазированы, часто не опускаются в мошонку, иногда диагностируется анорхия – полное отсутствие ткани яичек в организме. Наблюдается сильно заниженный уровень мужского гормона – тестостерона.
Источник
Содержание
- Синонимы диагноза
- Описание
- Дополнительные факты
- Причины
- Патогенез
- Симптомы
- Возможные осложнения
- Диагностика
- Лечение
- Список литературы
Другие названия и синонимы
Полная дисгенезия гонад, Чистая дисгенезия гонад.
Названия
Название: Синдром Свайера.
Синдром Свайера
Синонимы диагноза
Полная дисгенезия гонад, Чистая дисгенезия гонад.
Описание
Это нарушение формирования пола, характеризующееся кариотипом 46XY, врожденным дисгенезией гонад при первом образовании других женских половых органов — влагалища, матки, маточных труб. Признаки патологии включают первичную аменорею, мужское телосложение, сексуальный инфантилизм. Диагноз ставится на основании данных анамнеза, результатов общего и гинекологического обследования, интроскопических методов исследования органов малого таза, гормонального и молекулярно-генетического анализа. Лечение состоит из двух этапов: удаление неразвитых половых желез и длительное применение гормонозаместительной терапии.
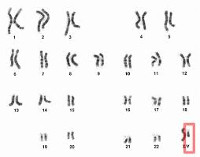
Синдром Свайера
Дополнительные факты
Синдром Свайера (полный или «чистый» дисгенезия гонад) можно кратко описать как женский фенотип в мужском генотипе. Болезнь названа в честь британского эндокринолога Джеральда Свиера, который описал ее в 1955 году как случай псевдогермафродитизма у мужчин. Полная форма дисгенеза не является синдромальной (не сопровождается экстрагенитальными пороками развития), исключает двойственность полового развития (наличие первичных мужских половых признаков с таковыми у женщин), психологическое развитие происходит по женскому типу. Врожденная патология встречается в одном случае у 20 000 человек с мужским кариотипом и регистрируется чаще, чем другие формы дисгенезии XY гонад.
Причины
Этиология синдрома Суайера не совсем понятна. На сегодняшний день известно, что появление патологии чаще всего связано с отсутствием или мутацией гена SRY, расположенного на коротком плече Y-хромосомы и главным образом ответственного за контроль образования семенников. Судя по наблюдениям за семейными случаями заболевания, возможно участие неизвестных Х-связанных или аутосомных генов.
Факторы риска также не полностью установлены. В дополнение к общим мутагенным эффектам (ионизирующее излучение и отравление, вирусные инфекции, несбалансированное или сниженное питание) и упомянутой выше наследственной нагрузке предполагается, что вероятность патологии может напрямую зависеть от возраста отца. Чаще всего невозможно проследить связь между каким-либо воздействием на организм во время беременности и развитием синдрома Свайера.
Патогенез
Формирование половых органов происходит из протока Мюллера у женщин, а у волка — у мужчин. В эмбрионе с мужским генотипом синтез мужских стероидных гормонов клетками Лейдига в эмбриональных яичках обусловлен действием хорионического гонадотропина матери. Клетки Сертоли стимулируют дифференцировку клеток Лейдига и других клеток, продуцируют гормон анти-Мюллера, который способствует атрофии протока Мюллера. Их нормальная активность приводит к развитию самца — с адекватной дифференцировкой яичек, атрофией протока Мюллера.
Отмеченные сбои этого механизма приводят к образованию женских репродуктивных органов из бипотентных почек, развитие которых не требует столь сложной регуляции. Созревание мужского зародыша контролируется геном SRY. При его отсутствии или мутации активность клеток Сертоли нарушается, дифференциация гонад не происходит, что влечет за собой развитие синдрома Свайера — фенотипически женского тела без полных яичников, которое может дополнительно стимулировать развитие вторичных половых признаков, но с бесполезным эмбрионы железы подвержены злокачественному новообразованию.
Симптомы
В допубертатном периоде патология продолжается без субъективных проявлений. В период полового созревания синдром Свайера характеризуется отсутствием признаков полового созревания. Наблюдается только редкий рост волос в лобковой и подмышечной областях, но он обычно отсутствует. Менархе не происходит, молочные железы не развиваются или выражаются очень слабо. Тип телосложения для дисгенеза яичников мужской — с широкими плечами, объемной грудью, узким тазом.
У женщин с синдромом Свайера чаще наблюдаются нормальный рост или рост выше среднего, развитые мышцы и «тяжелая» нижняя челюсть. Иногда возникает легкая гипертрофия клитора, хотя внешние половые органы обычно несколько недоразвиты. Пациенты жалуются на первичное бесплодие, чувство дискомфорта или боли из-за недостаточного развития влагалища во время полового акта или гинекологического осмотра.
Возможные осложнения
Распространенным (у 20-60% пациентов) осложнением синдрома Суайера является развитие опухолей, большинство из которых происходят из первичного полового отдела позвоночника, который на самом деле представляет собой дисгенетические гонады — дисгермин и андробласт. Эти новообразования часто носят злокачественный характер, встречаются с обеих сторон (синхронно или метахронно), регистрируются в раннем возрасте, в подростковом и детском возрасте.
Последствия нелеченного синдрома Суайера включают раннее появление патологий, вызванных дефицитом эстрогена у женщин — остеопороз, сердечно-сосудистые заболевания (гипертония, ишемическая болезнь сердца). Кроме того, для многих пациентов диагноз становится источником серьезных психологических страданий из-за «потери женственности», а для замужних женщин — и страха распада семьи.
Диагностика
Диагноз синдрома Сваера проводится гинекологом с участием медицинского генетика. Чаще всего диагноз ставится в возрасте от 14 до 15 лет, если вы обращаетесь к врачу по причине недостаточного полового развития, иногда позже, из-за бесплодия. В некоторых случаях первым признаком заболевания и основанием для углубленного обследования является образование опухоли из остаточных желез, которое было случайно обнаружено врачом или самим пациентом. Диагностические мероприятия включают в себя:
• Клиническое обследование. В ходе общего исследования отсутствие основных вторичных женских половых признаков определяется на фоне нормального или ускоренного роста. Гинекологическое обследование показывает нормальные, но недоразвитые женские наружные половые органы. Косвенным подтверждением диагноза является наличие кровных родственников с полным или частичным дисгенезией гонад.
• Методы радиационных исследований. Наиболее доступным и информативным методом диагностики является гинекологическое УЗИ. Ультрасонография позволяет обнаружить объективные характеристики, характерные для патологии, такие как гипоплазия матки, недоразвитые и осложненные маточные трубы и тяжелые яичники (иногда с новообразованиями). В сомнительных случаях назначается МРТ.
• Гормональный анализ. Характерным признаком синдрома Свиера является значительное увеличение гонадотропных гормонов (фолликулостимулятор, лютеинизатор) в сыворотке крови и снижение уровня эстрадиола. Гестагенный функциональный тест отрицательный, циклический гормональный тест (эстрогенно-гестагенный) положительный.
• Генетический тест. Он используется для проверки диагноза. Цитогенетическое исследование выявляет мужской кариотип (46, XY) и молекулярную генетику — отсутствие или повреждение гена SRY. Если генетическое тестирование невозможно, назначается гистологическое исследование биопсии яичника. Диагноз подтверждается структурой соединительной ткани без фолликулов.
Синдром Свайера отличается от других форм дисгенезии гонад — синдром Шерешевского-Тернера, мозаицизм 45, X / 46, XY, синдром Морриса (синдром феминизации яичек), а также с задержкой полового развития гипоталамо-гипофизарного генеза конституциональный и идиопатический. Если есть подозрение на опухоль, необходимо проконсультироваться с онкогинекологом и эндокринологом для выявления центральных форм позднего полового созревания.
Лечение
Лечение заболевания направлено на предотвращение осложнений, нормализацию психоэмоционального статуса пациента и, если возможно, достижение репродуктивной функции. После постановки диагноза проводится операция (удаление рудиментарных половых желез), затем назначается гормональная терапия. Раннее начало лечения синдрома Свайера (с подросткового возраста) повышает вероятность рождения ребенка.
• Заместительная гормональная терапия. Это осуществляется с помощью чередующихся препаратов эстрогена и прогестагена во время менструального цикла. Это предписано от половой зрелости до возраста менопаузы. Лечение нормализует течение обменных процессов, способствует развитию матки, женских половых вторичных признаков и гравидарной подготовки эндометрия.
• Коррекция нервно-психических расстройств. Психотерапевтическое воздействие на личность позволяет реконструировать отношение пациента к себе и своему непосредственному окружению, трансформировать его психологические установки и снять психоэмоциональный стресс. Иногда лечение сочетается с фармакотерапией (антидепрессанты, транквилизаторы).
При достаточном развитии матки можно использовать программу экстракорпорального оплодотворения с имплантацией донорских ооцитов, оплодотворенных спермой супруга пациента. Во время вынашивания плода женщине сначала прописывают поддерживающую, а затем консервативную гормональную терапию. Доставка производится в кратчайшие сроки. Успешный опыт ведения беременности у пациентов с дисгенезией гонад (включая синдром Свиера) уже накоплен в современной репродукологии и акушерстве.
Хирургическое вмешательство в объеме удаления рудиментарных половых желез и маточных труб выполняется сразу после постановки диагноза этой формы врожденного агенеза яичников. Хирургическое лечение проводится для предотвращения новообразований, источником которых являются клетки дисгенетических половых желез. Консервативное лечение не рекомендуется начинать до операции, так как гормональная терапия повышает риск развития раковых осложнений.
Список литературы
1. Аномалии половых хромосом при нарушениях формирования пола и репродукции человека. Автореферат диссертации/ Черных В. Б. – 2015.
2. Генетика человека/ Фогель Ф. , Мотульски А. – 1990.
3. Гинекология: национальное руководство/ под ред. Кулакова В. И. , Савельевой Г. М. , Манухиной И. Б. – 2009.
Источник