Ди джорджи синдром мкб 10

Рубрика МКБ-10: D82.1
МКБ-10 / D50-D89 КЛАСС III Болезни крови, кроветворных органов и отдельные нарушения, вовлекающие иммунный механизм / D80-D89 Отдельные нарушения, вовлекающие иммунный механизм / D82 Иммунодефициты, связанные с другими значительными дефектами
Определение и общие сведения[править]
Синдром Ди Джорджи
Синонимы: велокардиофациальный синдром, синдром делеции 22q11.2-хромосомы, синдром Такао, синдром Седлакова, синдром Шприцена.
Синдром Ди Джорджи (СДД) — изолированный Т-клеточный иммунодефицит. Характеризуется триадой ведущих клинических проявлений: аплазия или гипоплазия тимуса и/или паращитовидных желёз и врождённым пороком сердца.
Этиология и патогенез[править]
В основе синдрома Ди Джорджи лежит порок развития третьего и четвёртого глоточных карманов, возникающий между 6-й и 10-й неделями гестации и реализующийся в отсутствие или дисгенезию паращитовидных желёз и тимуса, а также тяжёлые пороки сердечно-сосудистой системы.
Болеют как мальчики, так и девочки, мутации в области 22q11.2 встречаются с частотой 1:3000-1:6000.
Патогенез
В большинстве случаев у больных обнаруживается микроделеция специфических последовательностей ДНК в области 22q11.2. В результате мутации стволовые клетки не дифференцируются в Т-лимфоциты. Синдром Ди Джорджи представляет собой изолированный Т-клеточный дефицит, при котором реакции клеточного иммунитета не определяются. При этом дети сохраняют способность к выработке специфических антител на очень низком уровне. Пациенты обладают сопротивляемостью к бактериальным инфекциям, но вирусные инфекции, такие как корь, ветряная оспа, протекают в тяжёлой форме. Вовлечение первого и второго жаберных карманов приводит к пороку развития лицевых структур, а заинтересованность пятого кармана проявляется широким спектром врождённых пороков сердца с частым вовлечением дуги аорты.
Клинические проявления[править]
У большинства больных отмечаются диспластические черты лица. Наиболее характерны диспластичные ушные раковины, гипертелоризм, широкая переносица, «рыбий рот», антимонголоидный разрез глаз. У части детей наблюдаются и более грубые аномалии, такие как микрогнатия и незаращение твёрдого и мягкого нёба.
Врождённые пороки сердца и магистральных сосудов также относятся к наиболее характерным и тяжёлым признакам заболевания (наиболее часто встречаются тетрада Фалло, декстрапозиция аорты, аномальная поключичная артерия).
Гипокальциемия различной степени тяжести и отсутствие тени вилочковой железы при рентгенографии грудной клетки относятся к частым проявлениям. Гипокальциемические судороги обычно возникают с первых дней жизни.
Характерна задержка физического и психомоторного развития. В раннем возрасте отмечается беспокойство, психоэмоциональная лабильность. В подростковом и взрослом возрасте больных СДД отмечаются психические отклонения, такие как шизофрения, маниакально-депрессивный психоз.
Клинические признаки иммунной недостаточности проявляются после рождения в виде повторных инфекций. Вирусные инфекции, такие как парагрипп, ротавирус, аденовирус, протекают у пациентов с синдромом Ди Джорджи в тяжёлой форме. При снижении Т-лимфоцитов ‹500 клеток в 1 мкл повышается риск присоединения оппортунистических инфекций (системных грибковых, пневмонии, вызванной Pneumocystis jiroveci) и клиника может напоминать ТКИН.
Задержка созревания и развития регуляторных функций Т-клеток приводит к развитию Т-клеток, обладающих аутореактивностью. В связи с этим у больных с синдромом Ди Джорджи отмечаются аутоиммунные заболевания: ювенильный ревматоидный артрит, аутоиммунная гемолитическая анемия, тромбоцитопения, аутоиммунный тиреоидит.
Отмечается вариабельность клинических проявлений у конкретных больных синдромом Ди Джорджи, у которых выделяются частичные формы синдрома. У части этих детей сохранена паратиреоидная функция, широко варьирует тяжесть врождённых пороков сердца (вплоть до полного отсутствия).
Иммунная недостаточность, за редким исключением, не определяет прогноз и спектр ведущих клинических проявлений при СДД. В большинстве случаев, если пациент переживает 6-месячный возраст, наблюдается постепенное спонтанное восстановление Т-клеточного иммунитета
Синдром Ди Джорджи: Диагностика[править]
1. Рентгенологическое исследование органов грудной клетки (прямая и боковая проекции) или МРТ: отсутствует тень вилочковой железы.
2. УЗИ сердца, инвазивные методы исследования сосудов с целью уточнения сердечно-сосудистой патологии.
3. Иммунологический спектр: количественные показатели Т-клеток варьируют от нормы до глубокой депрессии. Характерна диссоциация между сниженными уровнями Т- и НК-клеток и повышенным содержанием В-лимфоцитов. Характерны нормальные или повышенные уровни антител. Иммунный статус больных может эволюционировать как в сторону нормализации, так и более глубокой депрессии.
4. Гипокальциемия (связанная с низким уровнем паратгормона).
Дифференциальный диагноз[править]
Дифференциальная диагностика включает в себя синдром Смита-Лемли-Опица, синдром CHARGE, синдром Алажиля, синдром VATER, синдром Гольденхара и изотретиноиновую эмбриопатию.
Синдром Ди Джорджи: Лечение[править]
Коррекция Т-клеточных нарушений при синдроме Ди Джорджи может быть достигнута трансплантацией фетального тимуса. При наличии тяжёлых пороков, в основном определяющих прогноз для жизни, пересадка тимуса считается недостаточно обоснованной.
При наличии инфекционных проявлений показано проведение антибактериальной терапии антибиотиками широкого спектра действия. В случае рецидивирующей молочницы показан приём противогрибковых препаратов (низорал, дифлюкан, орунгал).
Применение гормонов тимуса для коррекции иммунных изменений не даёт существенного эффекта.
Прогноз
Прогноз зависит от степени выраженности гемодинамических расстройств и иммунодефицита. По данным литературы, летальность на первом месяце жизни составляла 55%, а летальность в первые 6 месяцев жизни составляла 86%. Причинами являлись пороки сердца и инфекции. При полных формах синдрома Ди Джорджи прогноз неудовлетворителен, течение заболевания приближается к ТКИН, частичные формы нередко с возрастом компенсируются.
Профилактика[править]
Прочее[править]
Источники (ссылки)[править]
Аллергология и иммунология [Электронный ресурс] / Под ред. Р.М. Хаитова, Н.И. Ильиной — М. : ГЭОТАР-Медиа, 2009. — https://www.rosmedlib.ru/book/ISBN9785970409039.html
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
Источник
Общие сведения
Синдром Ди Джорджи еще в некоторых источниках указывают как синдром Ди Георга. Впервые патология была описана в 1965 году американским педиатром и эндокринологом — Анджело Мария Ди Джорджи (фото). По международной классификации десятого пересмотра (МКБ-10) данному иммунодефициту был присвоен код D82.1 и указан как синдром дивертикула глотки, вилочковой железы: алимфоплазия, аплазия либо гипоплазия с иммунной недостаточностью.
")
Angelo DiGeorge(15.04.1921-11.10.2009)
Синдром Ди Джорджа как первичный иммунодефицит является редким врожденным заболеванием (1 случай на 4-6 тыс. человек) и относится к идиопатическому изолированному гипопаратиреозу – состоянию, для которого характерно снижение выработки паратгормонов и гипокальциемия. Для синдрома свойственно множество фенотипов поражения, осложнений и присоединения сопутствующих нарушений.
Патогенез
Для синдрома Ди Георга характерна аплазия или гипоплазия вилочковой железы (тимуса), а также агенезия или дисгенезия паращитовидных желез в результате нарушений закладки и эмбриональной дифференцировки 3-4 жаберных (глоточных) карманов. Это вызывает резкое снижение популяции Т-лимфоцитов, нарушение их дифференцировки и как следствие — иммунологическую недостаточность. У детей могут развиваться различные врождённые аномалии крупных сосудов, например, дефекты аорты, межжелудочковой перегородки или синий порок сердца.

Классификация
В зависимости от спектра клинических проявлений и врожденных аномалий синдром Ди Георга может быть полным и частичным, к примеру, протекать как изолированная недостаточность паращитовидных желёз либо врождённое отсутствие паращитовидных (околощитовидных) желез, которые приводят к гипокальциемическим судорогам, наблюдающимся у новорожденных в виде неонатальной тетании. Нарушения закладки тимуса приводят к развитию различных инфекционных заболеваний и обычно вызваны иммунологической недостаточностью.
Достаточно часто болезнь протекает в менее тяжелой форме и является наиболее распространенной причиной умственной отсталости с малым количеством других проявлений, поэтому может быть формально не диагностирована.
Причины
У синдрома Ди Джорджи или врождённой аплазии тимуса и паращитовидной железы генетическая причина развития. Патология развивается в результате делеции центрального участка более длинного плеча двадцать второй хромосомы, поэтому еще обозначается как синдром 22q 11.2. Однако, в некоторых случаях аналогичная клиническая картина наблюдалась и после других хромосомных перестроек – транслокаций и микроделеций, развивающихся в таких хромосомах как ТВХ1, 10р13, 17р13, 18q21 и пр. Эти мутации спорадические в 90% и обычно происходят на этапе мейоза спермато— или овогенеза.
Для данного синдрома дисэмбриогенеза 3-4 жаберной дуги — фенотипа CATCH 22 характерно наследование по аутосомно-доминантному типу, которое наблюдается лишь в 5-10% случаев, но при этом не исключается возможность аутосомно-рецессивного типа наследования с разной экспрессивностью.
Симптомы врождённой аплазии тимуса и паращитовидных желёз (Синдром Ди Джорджи)
Синдром Ди Джорджи – это первичный иммунодефицит, вызванный врожденным отсутствием или наличием аномалий развития тимуса и паращитовидных желез, который отличается триадой клинических симптомов, включающей:
- различные врожденные пороки сердца, дефекты крупных сосудов, носа, рта, ушей, что в результате дает специфические черты лица (как на фото): гипертелоризм (увеличенное расстояние между глазами), микрогнатия (недоразвитие нижней челюсти), низкое расположение ушных раковин;
- первичный иммунодефицит – нарушен как клеточный, так и гуморальный ответ иммунитета в результате аплазии вилочковой железы и невозможности обеспечения нормального развития Т-клеток;
- прогрессирующая гипокальциемия (пониженная концентрация кальция в кровотоке) – вызвана гипопаратиреозом, сопровождается судорожным синдромом и развитием скелетных аномалий.
Ребенок со специфическими чертами, характерными для синдрома Ди Джорджи
Особенности синдрома могут широко варьироваться даже в пределах одной семьи и затрагивать различные системы органов. Сопутствующими проблемами становится:
- нарушение работы почек и их атрофия, развитие нефрокальциноза, гидронефроза и пр.;
- проблемы с пищеварением и перистальтикой ЖКТ;
- дефицит гормона роста;
- проблемы с речью;
- психические расстройства, в том числе шизофрения;
- потеря слуха (проводящая и нейросенсорная обычно вызвана черепно-лицевыми синдромами);
- нарушения координации, которые могут быть вызваны гипоплазией мозжечка;
- синюшность кожных покровов в результате плохого кровообращения;
- ревматоидный артрит;
- частые переломы костей;
- болезнь Грейвса;
- болезнь Паркинсона.
Анализы и диагностика
Для подтверждения диагноза синдром Ди Джорджа необходимо выявление агенезии или дисгенеза паращитовидных желёз, аплазии вилочковой железы, иммунологической недостаточности, черепно-лицевых дисморфий (микрогнатии, гипертелоризма, антимонголоидного разреза глаз, расщелины губы и нёба, деформированных и/или низко расположенных ушных раковин) и прочих типичных аномалий развития. При этом оказываются эффективными различные исследования, включая УЗИ, МРТ и пр., а также используются методы генетического тестирования, к примеру, FISH (метод флуоресцентной гибридизации in situ).
Наиболее яркими проявлениями является гипопаратериоз и молочница. Также при плановом обследовании выявляются дефекты аорты, тетрада Фалло, катаракта, паховая грыжа и тому подобное. При помощи серологических исследований можно выявить лимфоцитопению, гипокальциемию, гипоγ-глобулинемии. Благодаря иммунологическим методам удается обнаружить нарушения трансформации лимфоцитов и их функциональной активности, в 20% случаев наблюдается сниженное количество Т-клеток. После иммунологических исследований важно провести дифференциальный диагноз с прочими первичными иммунодефицитами, таким как: синдром Вискотта-Олдрича, Брутона.
Лечение
Так как недуг неизлечим больным обычно назначают симптоматическое и комплексное поддерживающее лечение. Основными приемами считается соблюдение диеты, стерильные условия, прием кальция, комплексов витамин, антибиотиков, противогрибковых, иммуномодулирующих, седативных препаратов и пр.
Доктора
Лекарства
В зависимости от особенностей течения и имеющихся проблем со здоровьем могут быть назначены:
- противовирусные, противогрибковые препараты, а также антибиотики широкого спектра действия для лечения инфекционных заболеваний;
- пожизненный прием витамина Д и кальция;
- заместительная терапия иммуноглобулином.
Процедуры и операции
Для наилучшего прогноза пациентов с синдромом Ди Георга необходимо раннее выявление всех аномалий развития и их устранение стандартными методами лечения:
- трансплантация эмбриональных тканей тимуса;
- кардиохирургия при врожденных пороках сердца.
Первичный иммунодефицит у детей
В результате клеточного иммунодефицита дети с синдромом Ди Джорджи очень подвержены действию инфекции вирусной, грибковой и некоторых разновидностей бактериальной природы. Чаще всего развивается затяжной и тяжелый ринит, пневмония, абсцесс, пиелонефрит, колит, сепсис и т.д. Если ребенку удается пережить пятилетний возраст, то может не обнаруживаться недостаточность Т-клеток, так как антиген-независимый этап созревания Т-клеток начинает происходить не в тимусе, а в многослойном плоском эпителии.
У новорожденных часто возникают судороги и проблемы с кормлением. Младенцы слабые, у них плохой аппетит. Нарушение перистальтики приводит к частым запором. В дальнейшем у деток наблюдается задержка роста и развития, возникают проблемы с обучением, а также когнитивные нарушения, например, приобретение аутистических черт поведения.
У детей с синдромом ДиДжорджа специфический профиль, выявляемый нейропсихологическими тестами. У них нормальный IQ и они способны проходить обучение в обычной школе, дома или заниматься в специальных классах, ведь очень часто у больных есть проблемы с речью (неразборчивость, ошибки артикуляции, сложности при приобретении словарного запаса).
Прогноз синдрома Ди Джорджи
К сожалению, не сегодняшний день, современная медицина оказывается бессильна перед данным генетическим заболеванием. Прогноз крайне неблагоприятный – дети умирают от различных инфекционных заболеваний или сердечной недостаточности, поэтому лучшим способом предупреждения развития врожденных патологий считается медико-генетическое консультирование и пренатальная диагностика наследственных болезней.
Список источников
- Стефани Д.В., Вельтищев Ю.Е. Иммунология и иммунопатология детского возраста.— М., 1996, -С. 27.
- Ивановская Т.Е., Цинзерлинг А.В. Патологическая анатоми.— М., 1976. -С. 136.
Источник
Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Дифференциальная диагностика
- Лечение
- Прогноз
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Велокардиофациальный синдром.
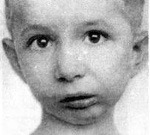
Велокардиофациальный синдром
Описание
Велокардиофациальный синдром. Врожденное (иногда наследственное) заболевание, характеризующееся множественными пороками развития и расстройством когнитивных функций. Его основными симптомами являются дефекты твердого неба (явные и скрытые расщелины), сердечно-сосудистые нарушения, задержка умственного развития, характерные черты лица. Диагностика велокардиофациального синдрома производится на основании настоящего статуса пациента (в том числе и при обследовании новорожденных) и данных цитогенетических исследований. Специфического лечения заболевания нет, применяют симптоматическую терапию, в том числе и хирургические методики.
Дополнительные факты
Велокардиофациальный синдром – генетическое заболевание, основа которого лежит в нарушении внутриутробного развития многих органов и систем организма. Существует несколько сходных фенотипов с подобными проявлениями (например, синдромы Ди Джорджи и Шпринтцена), которые, к тому же, обусловлены похожими генетическими причинами. Поэтому одни исследователи в области генетики объединяют все эти состояния в группу «велокардиофациальный синдром», тогда как другие полагают, что это все-таки различные нозологические единицы. Например, отличием болезни Ди Джорджи являются иммунологические нарушения, которые слабо выражены при велокардиофациальном синдроме. Встречаемость составляет около одного случая на 3000-4000 родов, с равной вероятностью развивается как у мальчиков, так и девочек, в большинстве случаев патология возникает спонтанно и не имеется у родителей или близких родственников ребенка. Лишь в отдельных наблюдениях надежно доказана наследственная передача велокардиофациального синдрома по аутосомно-доминантному механизму.
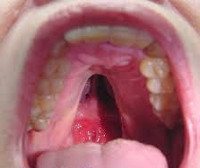
Велокардиофациальный синдром
Причины
Непосредственной причиной развития велокардиофациального синдрома являются нарушения структуры длинного плеча 22-й хромосомы – локуса q11. 2. Подобные дефекты возникают в процессе мейоза при образовании половых клеток, после чего передаются потомству. Чаще всего это микроделеции, которые сводятся к отсутствию небольшого участка хромосомы, при этом из генома больного исчезают важные гены, в частности TBX1. Роль этого гена в развитии велокардиофациального синдрома доказывает тот факт, что при некоторых формах заболевания отсутствует делеция участка 22-й хромосомы, но обнаруживаются точковые мутации в TBX1. Этот ген является одним из факторов регуляции транскрипции, наиболее активно участвующим в процессах эмбрионального развития.
Нарушение структуры TBX1 или тем более отсутствие в геноме в результате делеции приводит к патологиям эмбриогенеза, что становится причиной врожденных пороков развития. В частности, нарушаются процессы миграции клеток в области четвертого жаберного кармана и нервного гребня. По этой причине при велокардиофациальном синдроме затрагиваются именно те структуры, которые развиваются из данных эмбриональных зачатков. Сильнее всего это отражается на лице, сердечно-сосудистой, нервной и эндокринной системах. У больных велокардиофациальным синдромом возникают многочисленные нарушения в вышеуказанных системах органов, которые требуют длительного и иногда хирургического лечения. Иногда поражаются и другие структуры, не относящиеся к этим эмбриональным зачаткам – почки, мочеточники и мочевой пузырь, половые железы (крипторхизм). Такие проявления при велокардиофациальном синдроме обусловлены либо вторичными процессами, либо же тем обстоятельством, что при делеции могут повреждаться и другие гены помимо TBX1.
Делеция длинного плеча 22-й хромосомы или мутации гена TBX1 часто возникают в половых клетках родителей, то есть не встречаются у родственников пациента. Однако описаны случаи с аутосомно-доминантным механизмом наследования, по этой причине если один из родителей страдает велокардиофациальным синдромом, то вероятность рождения ребенка с данной патологией составляет 50%. Подмечено, что сходные с данным заболеванием синдромы Ди Джорджи и Шпринтцена чаще возникают не спонтанно, а именно передаются по наследству. И в этом случае механизм передачи нарушений аутосомно-доминантный.
Симптомы
Проявления велокардиофациального синдрома можно обнаружить уже сразу при рождении ребенка, однако это не всегда позволяет сразу же поставить такой диагноз. Во-первых, это связано со схожестью проявлений заболевания с другими аналогичными синдромами (Ди Джорджи, Шпринтцена), во-вторых, часть симптомов начинает проявляться с возрастом и не регистрируется в младенчестве. Одним из наиболее явных проявлений велокардиофациального синдрома является характерный тип лица – выявляется отведение нижней челюсти назад, малый размер носа и рта, расширение переносицы. Могут наблюдаться деформации хрящей носа и ушных раковин. Расщелина твердого нёба присутствует у всех больных велокардиофациальным синдромом, но выраженность этого нарушения может быть различна – от резко явного до почти незаметного, выявляемого только при инструментальных исследованиях.
Помимо этого, уже в младенческом возрасте могут наблюдаться судороги, обусловленные низким уровнем кальция в крови по причине гипофункции паращитовидных желез. Их необходимо дифференцировать от судорожных припадков, обусловленных поражением ЦНС, что также может иметь место при велокардиофациальном синдроме – однако, чаще всего они возникают намного в более позднем возрасте. Из других неврологических нарушений могут встречаться задержка психического развития, психические заболевания. Нарушения речи могут иметь как неврологическую природу, так и быть обусловлены пороками развития нёба и лицевого скелета. Почти у четверти больных наблюдается недобор массы тела в грудном периоде, с возрастом практически у всех пациентов отмечается отставание в физическом развитии той или иной степени выраженности.
Диагностика
Диагностика велокардиофациального синдрома производится на основании данных настоящего статуса пациента, изучения сердечно-сосудистой, эндокринной и нервной систем, а также генетических исследований. При этом для подтверждения диагноза без участия врача-генетика может потребоваться несколько лет наблюдений, так как многие из симптомов неспецифичны, а некоторые проявляются только по достижении больными определенного возраста. При осмотре новорожденного выявляют расщелину нёба той или иной степени выраженности, иногда она сопровождается и расщеплением верхней губы. У больных велокардиофациальным синдромом также определяется характерный внешний вид – широкая переносица, выступающий нос, маленький рот, низкорослость. У младенцев довольно часто обнаруживается недобор массы тела, а в дальнейшем – отставание в физическом развитии от сверстников. При пальпации может определяться мышечный гипотонус, заторможенность рефлексов.
Изучение функций сердечно-сосудистой системы при помощи ЭхоКГ почти в 70% случаев выявляет те или иные аномалии развития, которые ассоциируются с этим заболеванием. Это могут быть дефекты межжелудочковой перегородки, тетрада Фалло, необычное расположение артериальных сосудов. В редких случаях может выявляться такое состояние, как дестрокардия. Больным необходимо регулярное диспансерное наблюдение у врача-кардиолога, так как ряд нарушений может возникать или прогрессировать со временем. При развитии судорог необходимо провести анализ крови для определения уровня кальция и паратгормона – оба этих показателя могут быть значительно снижены. Изменение функций других желез внутренней секреции, как правило, не определяется.
Важным аспектом диагностики и лечения велокардиофациального синдрома является наблюдение за психофизическим развитием ребенка. В большинстве случаев в раннем детском возрасте больные дети значительно отстают от своих сверстников, медленнее растут, позже начинают говорить, имеют затруднения в усвоении новой информации. Это может иметь как первичные неврологические причины, так и быть обусловлено вторичными обстоятельствами – расщелиной твердого нёба (затрудняется питание новорожденных и речь у более старших детей), мышечным гипотонусом, проблемами с сердечно-сосудистой системой (хроническое кислородное голодание тканей). Однако при своевременной коррекции нарушений отставание в развитии при велокардиофациальном синдроме уменьшается к школьному возрасту.
Генетическая диагностика велокардиофациального синдрома включает в себя выявление участков микроделеций на длинном плече 22-й хромосомы и поиск точковых мутаций в гене TBX1. Так как делеции встречаются статистически чаще, то генетическое подтверждение заболевания начинают именно с их выявления. Для этой цели чаще всего используют различные варианты мультиплексной полимеразной цепной реакции. В отношении гена TBX1 применяют метод прямого секвенирования последовательности с целью определения дефектных участков.
Дифференциальная диагностика
Дифференциальную диагностику велокардиофациального синдрома производят с другими патологиями, обусловленными микроделециями 22-й хромосомы (синдромы Ди Джорджи и Шпринтцена) и синдромом «кошачьего крика».
Лечение
Специфического лечения велокардиофациального синдрома не существует, возможно только симптоматическое лечение и коррекция аномалий развития нёба, лица, сердечно-сосудистой системы. Самой частой паллиативной операцией при этом заболевании является коррекция расщелины твердого неба – нередко ее производят еще в младенческом возрасте, так как этот дефект значительно осложняет питание ребенка. Судороги, обусловленные гипокальциемией, устраняют назначением препаратов кальция – в большинстве случаев это позволяет полностью купировать данное проявление велокардиофациального синдрома. В ряде случаев необходима хирургическая коррекция аномалий сердца и сосудов, которая может производиться кардиохирургами в различном возрасте в зависимости от характера, выраженности и других обстоятельств.
Важным аспектом лечения велокардиофациального синдрома является помощь в психофизическом развитии ребенка. При гипотонусе мышц с раннего возраста показана физиотерапия и лечебная физкультура для улучшения координации движений и активности мышечных тканей. Особой проблемой при велокардиофациальном синдроме является развитие речи больного – она нарушена как из-за неврологических нарушений, так и по причине наличия расщелины твердого нёба. Поэтому хирургическое устранение этого дефекта и работа с логопедом необходимы для дальнейшей социализации ребенка. В ряде случаев требуется работа с психологом, коррекционным педагогом и вспомогательное обучение, часть детей со временем может посещать обычные школы и классы. У некоторых больных велокардиофациальным синдромом выявляется синдром дефицита внимания и гиперактивности (СДВГ), что требует применения дополнительных лекарственных и психотерапевтических мер.
Прогноз
Прогноз заболевания часто неопределенный по причине широкого спектра выраженности симптомов – в особенности аномалий развития сердечно-сосудистой системы. При тяжелых формах велокардиофациального синдрома пороки развития сердца и сосудов могут приводить к смерти больного в раннем возрасте. Однако в большинстве случаев своевременная хирургическая коррекция и симптоматическое лечение значительно повышают как выживаемость пациентов, так и качество жизни. Так как данная патология является комплексной, то для контроля за ее течением и устранения нарушений нужна помощь множества медицинских специалистов. Необходимо регулярное посещение кардиолога, работа с психологом и логопедом, лечебная физкультура – все это позволит заметно снизить выраженность проявлений велокардиофациального синдрома.
Основные медуслуги по стандартам лечения | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Клиники для лечения с лучшими ценами
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Источник