Дети синдром прадера вилли фото

Что такое синдром Прадера — Вилли?
Синдром Прадера — Вилли (СПВ) — это генетическое заболевание, которое встречается примерно у одного из каждых 15 000 новорожденных.
Синдром Прадера — Вилли поражает мужчин и женщин с одинаковой частотой и затрагивает все расы и этнические группы. СПВ признана наиболее распространенной генетической причиной опасного для жизни детского ожирения.
Данный синдром был впервые описан швейцарскими врачами Андреа Прадером, Алексисом Лабхартом и Генрихом Вилли в 1956 году на основании клинических характеристик девяти обследованных детей.
Общие характеристики, определенные в первоначальном отчете, включали маленькие руки и ноги, аномальный рост и состав тела (маленький рост, очень низкая мышечная масса тела и раннее детское ожирение), мышечная гипотония (низкий мышечный тонус (слабость мышц)) при рождении, ненасытный голод, сильное ожирение и умственная отсталость.
Это видео даст краткое понимание проявлений болезни:
Синдром Прадера — Вилли является результатом аномалии хромосомы 15, и постановка точного диагноза сегодня основывается на генетических анализах.
Каковы симптомы синдрома Прадера — Вилли?
Симптомы синдрома Прадера — Вилли, вероятно, связаны с дисфункцией части мозга, называемой гипоталамус. Гипоталамус — это небольшой эндокринный орган у основания мозга, который играет важную роль во многих функциях организма, включая регулирование голода и сытости, температуры тела, боли, баланса сна и бодрствования, водного баланса, эмоций и фертильности.
Хотя считается, что дисфункция гипоталамуса приводит к появлению симптомов синдрома Прадера — Вилли, еще не ясно, как генетическая аномалия вызывает дисфункцию гипоталамуса.
У людей с данным синдромом симптомы со временем меняются. В целом, есть два основных этапа симптомов, связанных с СПВ:
Ранний период жизни
Младенцы с СПВ являются гипотоническими или «гибкими», с очень низким мышечным тонусом. Для них типичен слабый крик и плохой сосательный рефлекс. Детей с синдромом Прадера — Вилли обычно не могут кормить грудью и они часто нуждаются в искусственном вскармливании.
У детей могут быть проблемы с ростом и развитием, если трудности с кормлением не подвергаются тщательному мониторингу и лечению. По мере взросления этих детей сила и мышечный тонус в целом улучшаются. Моторные функции развиты, но, как правило, выполняются с задержкой.
Малыши, как правило, вступают в период, когда они могут легко начать набирать вес до того, как проявят повышенный интерес к еде.
Детство и взросление
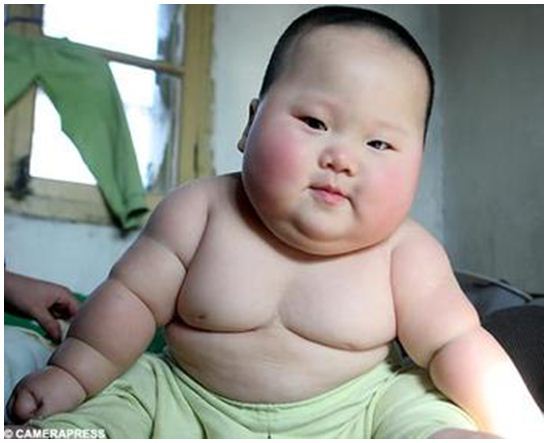
Нерегулируемый аппетит и легкое увеличение веса (см. фото) характеризуют более поздние стадии синдрома Прадера — Вилли.
Эти признаки чаще всего начинаются в возрасте от 3 до 8 лет, но различаются по началу и интенсивности.
Люди с синдромом Прадера — Виллии испытывают недостаток в нормальных подсказках голода и сытости.
Как правило, они не могут контролировать потребление пищи и переедают, если их не контролировать.
Поведение в голоде очень распространено. Кроме того, скорость метаболизма у людей с данным синдромом ниже, чем обычно. Без лечения эта комбинация проблем приводит к патологическому ожирению и его многочисленным осложнениям.
Помимо ожирения, у ребенка с синдромом Прадера — Вилли может быть множество других симптомов. Дети с IQ от низкого нормального до умеренной умственной отсталости обычно имеют когнитивные проблемы. Те, у кого нормальный IQ, обычно имеют проблемы с обучаемостью.
Другие проблемы и признаки могут включать:
- дефицит гормона роста/низкий рост;
- маленькие руки и ноги;
- сколиоз;
- нарушения сна с чрезмерной дневной сонливостью;
- высокий болевой порог;
- апраксия/диспраксия речи и бесплодие.
Поведенческие трудности могут включать в себя обсессивно-компульсивные симптомы, проблемы с кожей и трудности с контролем эмоций. Взрослые с синдромом Прадера— Вилли подвергаются повышенному риску психических заболеваний. СПВ является расстройством широкого спектра, и симптомы варьируют по степени тяжести и частоты встречаемости среди людей.
Хромосомы и гены: основы
Чтобы понять генетику СПВ, необходимо иметь общее представление о хромосомах и генах.
Хромосомы представляют собой крошечные структуры, которые присутствуют почти в каждой клетке нашего тела. Это пакеты генов, которые мы наследуем от наших родителей. Гены содержат все подробные инструкции, необходимые нашему телу для роста, развития и функционирования — нашу ДНК.
Определенные гены направляют наши клетки на производство белков, ферментов и других необходимых веществ. Каждый из наших многочисленных генов находится на определенной хромосоме. Большинство клеток нашего тела содержат 46 хромосом — 23 унаследованы от нашей матери и 23 от нашего отца. (Яйца и сперматозоиды обычно содержат всего 23 хромосомы, потому что это те клетки объединяются в зачатии и обеспечивают ребенку нужное количество хромосом.)
22 пары хромосом помечены числом, основанным на их размере (хромосома 1 — самая большая пара, а хромосома 22 — почти наименьшая), и 2 хромосомы в каждой пронумерованной паре содержат одинаковые гены (один набор у матери и одну от отца). Изменения, которые вызывают синдром Прадера — Вилли, происходят в паре, известной как хромосома 15. 23-я пара хромосом обозначена как пара половых хромосом. Эта пара определяет пол ребенка: XX для девочки, XY для мальчика.
Изменения или ошибки в генах и хромосомах распространены при образовании яйцеклеток и сперматозоидов. Некоторые из этих генетических изменений не будут иметь эффекта, когда ребенок зачат; некоторые вызовут выкидыш; а некоторые спровоцируют, например, синдром Прадера — Вилли, вызвав значительные различия в том, как ребенок развивается и функционирует.
В то время как многие генетические нарушения вызваны изменением одного гена и могут передаваться от родителя к ребенку, СПВ куда более сложен.
Что вызывает синдром Прадера — Вилли (причины)?
Синдром Прадера — Вилли возникает в результате отсутствия активного генетического материала в определенной области хромосомы 15 (15q11-q13). Обычно люди наследуют одну копию хромосомы 15 от своей матери и одну от своего отца. Гены вызывающие СПВ активны только в хромосоме, полученной от отца.
При синдроме Прадера — Вилли генетический дефект, вызывающий неактивность хромосомы 15 от отца (отцовская хромосома 15), может возникнуть по трем следующим причинам:
- СПВ по исключению: чаще всего часть хромосомы 15, которая была унаследована от отца, отсутствует или удалена в этой критической области. Эта небольшая делеция встречается примерно в 70% случаев и обычно не выявляется при обычном генетическом анализе, таком как амниоцентез.
- СПВ по однородительской дисомии: еще около 30% случаев происходят, когда человек наследует две хромосомы 15 от своей матери и ни один от своего отца. Это тип наследование называется однородительской дисомией (UPD).
- СПВ по импрессии мутации: наконец, в очень небольшом проценте случаев (1-3%) небольшая генетическая мутация при синдроме Прадера — Вилли приводит к тому, что генетический материал отцовской хромосомы 15 хоть и присутствует, но неактивен.
Как эти генетические дефекты вызывают симптомы, наблюдаемые при синдроме Прадера — Вилли?
Хромосомы 15 одни из самых сложных областей генома человека. Хотя были достигнуты значительные успехи в понимании и характеристике генетических изменений, связанных с синдромом Прадера — Вилли, точный механизм, с помощью которого отсутствие функционального генетического материала приводит к симптомам, связанным с СПВ, не понятны.
Ученые активно изучают нормальную роль генетических последовательностей в области СПВ и то, как их потеря влияет на гипоталамус и другие системы организма.
Существуют ли различия в степени тяжести СПВ в зависимости от генетического подтипа?
Могут быть некоторые тонкие различия в признаках СПВ на основе генетического подтипа: например, лица с делециями могут быть светлокожими со светлыми волосами по сравнению с другими членами семьи и быть более восприимчивыми к припадкам и судорогам; а те, у кого есть проблемы с СПВ по однородительской дисомии, могут быть подвержены более высокому риску психических заболеваний в молодом подростковом возрасте.
В целом, однако, существует значительное совпадение между различными генетическими подтипами. Вероятно, что тысячи генов за пределами области СПВ, которые демонстрируют нормальные различия между индивидуумами, также вносят значительный вклад в вариабельность симптомов синдрома Прадера — Вилли между теми, у кого есть расстройство.
Как диагностируется СПВ?
Данный синдром диагностируется с помощью анализа крови, который выявляет генетические аномалии, специфичные для синдрома Прадера — Вилли — так называемый «анализ метилирования ДНК». Метод FISH (флуоресцентная гибридизация in-situ) идентифицирует синдром путем делеции, но не диагностирует другие формы СПВ.
Анализ на метилирование ДНК идентифицирует все типы этого синдрома и является предпочтительным методом исследования для диагностики. Если анализ на метилирование проводится первым, может потребоваться дополнительное исследование, чтобы определить, вызван ли СПВ отцовской делецией, UPD или импринтинговой мутацией.
В случаях, когда существует подозрение на наличие импринтинговой мутации, кровь также может быть взята у родителей.
Каковы риски синдрома Прадера — Вилли в будущем?
СПВ по исключению и UPD являются случайными явлениями и, как правило, не связаны с повышенным риском рецидива в будущих беременностях. В случае импринтинг-мутации синдром Прадера — Вилли может в семье повториться. Семьи с опасениями по поводу их риска СПВ должны поговорить с генетическим консультантом.
Есть ли лекарство от синдрома Прадера — Вилли?
В настоящее время нет никакого лечения от синдрома Прадера — Вилли, и большинство исследований на сегодняшний день было направлено на лечение определенных симптомов данного синдрома.
Для многих людей, затронутых этим расстройством, устранение некоторых из самых сложных аспектов синдрома, таких как ненасытный аппетит и ожирение, будет представлять собой значительное улучшение качества жизни и способности жить независимо.
Источник
С развитием молекулярно-генетических методов процесс диагностики различных заболеваний сильно упростился. Синдром Прадера — Вилли — врождённое заболевание наследственной природы, сопровождающееся большим количеством осложнений. Патология является частой причиной ожирения у детей.
Генетический характер синдрома Прадера — Вилли
Синдром Прадера — Вилли является генетическим заболеванием, возникающим из-за нарушений функционирования определённого района 15-й хромосомы, унаследованной от отца. Гены в нём могут частично не работать, быть удалёнными или нести в себе только материал, полученный матери.
В наше время заболевание очень часто неправильно диагностируется, в связи с чем в первые годы жизни ребёнок не получает адекватного и своевременного лечения.
Впервые синдром был описан швейцарскими педиатрами А. Прадером и Г. Вилли. Согласно статистике, заболевание встречается у одного из 10–20 тысяч новорождённых. Долгое время было сложно установить связь между наследственностью и причиной возникновения патологии. С развитием науки и появлением различных молекулярных методов тестирования учёные смогли точно идентифицировать генетическое заболевание у всех пациентов.
Наследственность как причина заболевания
Существовало много предположительных вариантов передачи заболевания. С помощью генетических анализов было установлено, что к нарушению приводит в 70% случаев микроделеция (выпадение участка хромосомы) или отсутствие активности генов участка 15 хромосомы, полученной от отца, а у 25% пациентов — материнская изодисомия (все две хромосомы получены от матери). Оставшаяся часть возникает из-за замены одних генов на другие в определённом участке 15 хромосомы или нарушения их функционирования (импринтинга).
Импринтинг определяет степень влияния генов на строение и функционирование организма в зависимости от родителя, от которого они были получены. Подобная ситуация характерна не для всего наследственного материала, а только для 1 процента.
Клинических различий между больными при этом не было выявлено.
Заболевание встречается одинаково часто среди мальчиков и девочек. Синдром Прадера — Вилли является главной причиной развития ожирения у детей до года. У больных снижен процесс расщепления жира и усилена его выработка.
Дети рождаются доношенными. По внешним данным бывает сложно определить сразу наличие генетического заболевания. Однако при детальном изучении обнаруживается очень много отклонений.
У детей до полутора лет снижен мышечный тонус, реакция в виде вскидывания рук при испуге (рефлекс Моро) и сосательно-глотательные рефлексы. Родители испытывают сильные проблемы при кормлении ребёнка. Часто встречается затруднённое дыхание.
Рефлекс Моро проявляется в норме у детей с самого рождения и пропадает через несколько месяцев. Во время осмотра врач хлопает вблизи от младенца и наблюдает за ним. В норме ребёнок сильно разводит ручки и растопыривает пальцы, после чего начинает быстро их сближать. Со стороны кажется, что малыш пытается обнять и прижать к себе кого-то.
Симптомы болезни
Пациенты с синдромом Прадера — Вилли страдают от дисфункции центрального органа, управляющего всей эндокринной системой — гипоталамуса, расположенного в головном мозге. Возникающее вследствие этого недоразвитие яичек и яичников приводит к нарушению выработки гормонов и недоразвитию внешних и внутренних половых признаков. У мальчиков этот процесс является причиной отсутствия одного или обоих яичек в мошонке, а у девочек в половине случаев отмечается уменьшенная недоразвитая матка. Снижение количества специфического белка-фермента тирозиназы приводит к слабому пигментированию волос, кожи и радужки глаза.
Крипторхизм — неправильное расположение яичек, при котором они не выходят в мошонку, а остаются внутри брюшной полости, под кожей на бедре, лобке, в паху.
В дальнейшем у ребёнка развивается постоянное чувство голода, еда не приносит насыщения. Именно с этого момента начинается развитие ожирения. Для этого заболевания характерно отложение жира в нижней части туловища, в области живота и бёдер.
Постепенно мышечный тонус у ребёнка восстанавливается и развивается до нормального уровня примерно к шести годам. Однако становятся сильно заметны внешние дефекты: стопы и кисти рук непропорционально маленькие, ноги искривлены, чрезмерно опущены веки, деформирован позвоночник, рост ниже средних показателей для этого возраста. Ребёнок быстро утомляется, часто находится в сонливом состоянии, отстаёт физически от своих сверстников. Часто развивается косоглазие из-за мышечных дефектов.
Психическое состояние больных зачастую нестабильно. Несмотря на общую дружелюбность, у них наблюдаются вспышки гнева, немотивированной агрессии, истерики. Дети с синдромом вредничают, упрямятся. У многих больных встречается невроз навязчивых состояний, проявления которого очень многообразны, начиная с выполнения бытовых дел и заканчивая страхами и фобиями. Некоторые дети страдают дерматиломанией — сдиранием кусочков кожи на своём теле. До 10% больных испытывают галлюцинации, параноидальные отклонения и страдают от депрессий.
В школьном возрасте особенно выделяется нарушение речи и снижение интеллектуального развития.
В 1992 году учёные провели изучение интеллекта больных с синдромом Прадера — Вилли. В норме коэффициент интеллектуального развития составляет 85–115 баллов. Только 5% пациентов преодолели этот порог и остались на низком или среднем уровне развития, остальные находились ниже 85 баллов. Большинство имело умеренно сниженный интеллект и 30% обладали умственной отсталостью.
Дети с данным синдромом обладают необычным восприятием, у них сильно развито воображение, они отлично воспринимают информацию визуально, хорошо запоминают слова, быстро читают, но при всём этом имеют проблемы с устным выражением мысли и речью. Воспринимать информацию на слух больные могут плохо, их логическое и математическое мышление находится на крайне низком уровне. Письменное выражение мыслей может даваться им с большим трудом.
Во взрослой жизни больные сталкиваются с множеством проблем. Подавляющее большинство не может иметь детей, недоразвитие половых желёз приводит к бесплодности и различным аномалиям анатомического строения репродуктивных органов. Многим ставится диагноз сахарный диабет. Во внешнем облике больных выделяется широкий нос, миндалевидные глаза, узкие губы. Цвет глаз, волос и кожи таких больных светлее, чем у ближайших родственников.
Истории детей с синдромом Прадера — Вилли — видео
Одним из страшных заболеваний, которое чаще проявляется у больных с синдромом Прадера — Вилли, является извращённое кроветворение (лейкемия). Уровень восстановления генетической информации в белых клетках крови (лимфоцитах) снижен до 67%. В связи с этим у больных чрезвычайно повышен риск онкологических патологий системы кроветворения.
Проблемы с костной тканью проявляются во всём организме. Зубы сильно подвержены кариесу, а сколиоз прогрессирует с возрастом. При этом люди с данным синдромом очень гибкие, суставы легко гнутся в разные стороны, не вызывая болезненных ощущений.
Методы диагностики
Первые признаки заболевания врач может обнаружить уже во время беременности: слабая подвижность плода, многоводие, неправильные типы предлежания (ягодичное, поперечное). Рекомендована консультация врача, который назначит генетический анализ. Для получения клеток будущего ребёнка используется прокол плодного пузыря (амниоцентез). При диагностике крови матери обнаруживается сниженный уровень гормона гонадотропина.
После рождения самым точным определением наличия синдрома является обнаружение аномалий 15 хромосомы путём молекулярно-генетических исследований.
Дети с синдромом Прадера — Вилли — фотогалерея
С 1993 года были выделены два вида признаков заболевания: основные и дополнительные.
Основные признаки:
- снижение мышечного тонуса у детей до года;
- трудности кормления новорождённых;
- быстрый набор жировой массы;
- характерный внешний вид;
- повышенный аппетит;
- отставание в интеллектуальном развитии;
- недоразвитие половых желёз.
Дополнительные признаки:
- слабая подвижность плода в период внутриутробного развития;
- сложности поведения;
- нарушения цикла сна;
- маленький рост, кисти и стопы;
- слабая пигментация волос, кожи и радужной оболочки глаз;
- плотная, тянущаяся слюна;
- проблемы с речью.
При синдроме Прадера-Вилли необходимо проведение дифференциальной диагностики со следующими заболеваниями:
- миопатией;
- спинальной амиотрофией;
- синдромом Опица — Фриаса;
- синдромом Лоуренса — Муна;
- синдромом Барде — Бидля;
- синдромом Альстрема;
- синдромом Коэна;
- адипозогенитальной дистрофией;
- синдромом Дауна.Дифференциальная диагностика синдрома Прадера — Вилли обязательно проводится с синдромом Дауна
С задачей дифференциальной диагностики полностью справляется анализ ДНК, без которого множество случаев синдрома Прадера — Вилли остаются невыявленными. Врач может поставить диагноз синдром Дауна, так как он более распространён и изучен. В обоих случаях наблюдаются ожирение и умственная отсталость.
Лечение синдрома
К сожалению, заболевание является неизлечимым и будет сопровождать больного всю жизнь. Однако при ранней диагностике и своевременном начале терапии можно существенно улучшить жизнь ребёнка и снизить выраженность клинических проявлений.
Основные методы лечения
Во многих случаях больным назначается приём гормона роста в связи с пониженным производством его в организме ребёнка. Терапия помогает усилить рост пациента и набор мышечной массы и в некоторой степени даже снизит аппетит.
В связи с недоразвитием яичек и яичников врач назначает гормоны для стимуляции полового развития. В случае крипторхизма назначают операцию для выведения яичек в мошонку.
Так как большинство больных страдают нарушениями сна и внезапной остановкой дыхания в определённые моменты, врачом рекомендуется использование специального медицинского аппарата. Устройство предотвратит подобное негативное состояние, создавая постоянную вентиляцию лёгких за счёт давления.
Специальное устройство поможет избежать остановки дыхания во сне
Для лечения нарушений обмена веществ назначают метаболические препараты.
Физиотерапия, спорт, массаж
С младенческого возраста больному ребёнку необходимо проходить сеансы физиотерапии и массажа для укрепления мышц, поддержания их в тонусе.
Рекомендовано плавание, так как этот вид спорта позволяет сделать нагрузки на позвоночник минимальными, но при этом укрепить все мышцы организма, препятствуя сколиозу.
Дополнительные спортивные кружки отлично помогут ребёнку держать мышцы в тонусе, контролировать вес и полноценно физически развиваться. Ребёнку подойдёт детский фитнес, ЛФК и другие секции.
Диетотерапия
В связи с тем, что основная проблема больных с синдромом Прадера-Вилли — ожирение, врачи уделяют усиленное внимание строгой диете. Стоит ограничивать жирную пищу и быстрые углеводы. В дошкольном возрасте допускается полноценный рацион для развития организма. Затем назначают диету с дефицитом калорий (до 1200 в день), дополнительно используется приём поливитаминов и минералов.
В случае наличия сильных проблем с кормлением у новорождённых приходится прибегать к питанию через желудочный зонд.
Так как аппетит у таких детей неконтролируем, они часто воруют и прячут еду. Родители должны уменьшить доступ к пище, использовать различные запирающие средства на холодильник и шкафы.
Необходимо исключить из питания мучную продукцию, сладости, жирную пищу. Рацион ребёнка должен быть насыщен овощами, фруктами, нежирным мясом и рыбой. Необходимо использовать при приготовлении блюд растительные масла — оливковое, грецкое, льняное.
Занятия со специалистами
Обязательным мероприятием при синдроме Прадера — Вилли является наблюдение невропатолога, особенно на первых годах жизни. Врач точно установит степень задержки развития и поможет снизить выраженность симптомов болезни.
Обязательным является посещение логопеда-дефектолога. Квалифицированный врач поможет структурировать речь, научить правильному произношению звуков.
Занятия с логопедом необходимы ребёнку с синдромом Прадера — Вилли
Педиатр-эндокринолог должен постоянно проверять гормональный фон ребёнка, вовремя корректировать приём препаратов и следить за диетой.
Прогноз лечения
Полностью вылечиться от синдрома Прадера-Вилли невозможно, однако при правильном проведении терапии качество жизни больных существенно улучшается.
Продолжительность жизни больных может достигать шестидесяти и более лет. Чаще всего летальные случаи происходят из-за последствий ожирения: диабета, остановки дыхания во сне, заболеваний почек и сердечно-сосудистой системы.
Существенно возрастает продолжительность жизни у больных с синдромом Прадера — Вилли в семьях, которые соблюдают принципы правильного питания и поддерживают подвижный образ жизни.
Вероятность рождения в семье больного ребёнка
Так как синдром является врождённым, то его профилактика может заключаться только в дородовом анализе наследственного материала клеток плода и последующей консультации врача-генетика.
Если у первого ребёнка диагностировано наследование обеих хромосом от отца или выпадение в одной из них определённого участка, то вероятность рождения на свет следующего малыша с аналогичной патологией составляет один процент. Однако если причиной синдрома Прадера — Вилли являлось замещение участка хромосомы другим генетическим набором, то вероятность увеличивается до 50%. Аналогичным случаем является наследование обеих хромосом от матери. Зная результаты анализов, можно разумно оценить все возможные риски рождения ребёнка с данным заболеванием.
Генетический анализ клеток плода — метод профилактики синдрома Прадера — Вилли
Несмотря на неизлечимость болезни, своевременное и качественное лечение поможет ребёнку поддерживать нормальный вес, подвижность и развиваться интеллектуально с помощью специальных методик.
- Автор: Екатерина Сергеевна
- Распечатать
Высшее образование в сфере биологии (СПбГУ, магистр биологии), специализация — генетика человека.
Оцените статью:
Источник