Дети с синдромом прадера вилли в россии

Синдром Прадера — Вилли — редкое генетическое заболевание, о котором в России минимально осведомлены даже врачи. Лечения пока что не существует. Мы поговорили с двумя мамами, у чьих детей диагностировали синдром Прадера — Вилли, чтобы узнать, как живут с таким заболеванием в нашей стране и как они создали благотворительный фонд помощи людям с синдромом Прадера — Вилли.
Рассылка «Мела»
Мы отправляем нашу интересную и очень полезную рассылку два раза в неделю: во вторник и пятницу
«Самая большая опасность — это смерть от удушья из-за ожирения и задержек дыхания во сне»
Марина Амулина, мама ребёнка с синдромом Прадера — Вилли, сооснователь благотворительного фонда помощи людям с СПВ
Моя беременность была сложная: сильнейший токсикоз и всевозможные боли. Но так было и с первым ребёнком, поэтому особого значения я не придала. Так же, как и в первый раз, в 37 недель сильно ухудшилось состояние плаценты и пуповины. Ребёнок намного меньше двигался. Но все данные КТГ были хорошими, поэтому врачи не паниковали.
Мне сильно помогла интуиция. Поскольку все анализы были в норме, после последнего обследования меня спокойно отправили ждать родов. Но через пару дней я поняла, что нужно ехать в роддом на внеплановое УЗИ. Будто уже все знала подсознательно. В этот же день меня положили рожать. Потом оказалось, что, промедлив еще один день, врачи могли не спасти ребёнка.

Фото из личного архива Марины Амулиной
Роды были сложными. Врачи посчитали чудом, что сын родился живым. Домой выписывали меня одну. Все спускались в лифте с новорожденными, а я — с пакетом вещей. Роберта перевезли в реанимацию, куда я могла приезжать лишь на час в сутки.
Сын не дышал самостоятельно ровно неделю после рождения. У него не было никаких рефлексов: не мог сосать, глотать, хватать ещё долгое время.Две недели не открывал глаза. Он не издавал никаких звуков, не плакал до трёх месяцев. Он был похож на тряпочку, истыканную аппаратами, поддерживающими в нём жизнь.
К месяцу он научился глотать сам, но делал это неправильно. Если вдруг капля еды попадала не туда, он не мог самостоятельно откашливаться. Соответственно, всё шло в лёгкие. А это опять реанимация. В итоге до трёх месяцев Роберт ел через желудочный зонд — специальную трубочку, подающую пищу в организм.
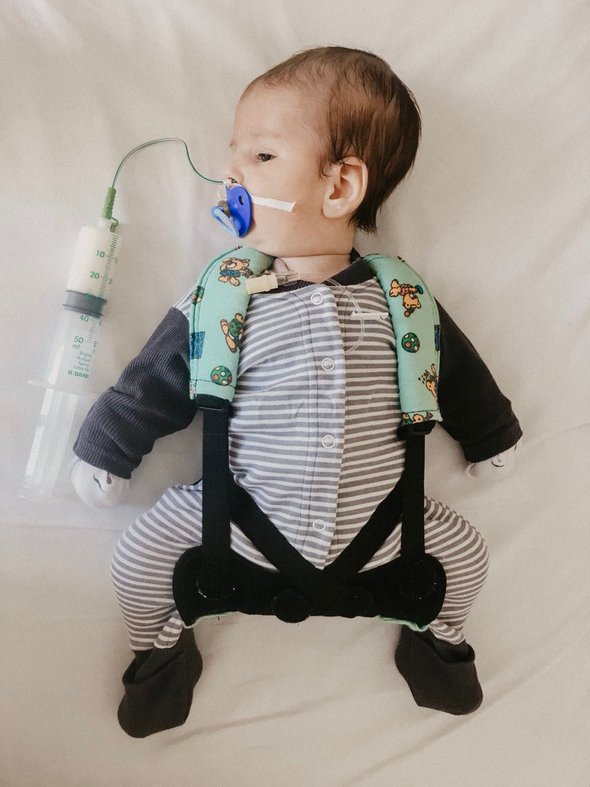
Фото из личного архива Марины Амулиной
Помимо этого, ночью у него часто случалось апноэ — задержка дыхания. Мне приходилось отлавливать эти моменты. При дыхании он очень сильно хрипел, все время. Его мышцы настолько слабы, что весь первый год он не мог самостоятельно удерживать голову. Сейчас Роберту два года, и до сих пор он не может правильно держать спину, не может ходить.
Проблемы были практически везде: мозг, зрение, суставы, сердце, почки, легкие, печень, половая система, центральная нервная система
Мы всё время лежали с малышом на обследованиях и реабилитациях, чтобы понять, что с ним. Мы посетили слишком многих московских врачей, потратили немало времени и денег, но никто не мог помочь нам понять, что с сыном, диагноз поставить не мог никто. Нам не назначали никакого особого лечения, потому что состояние всегда было довольно тяжелое. Любой неправильный шаг мог привести к эпилепсии. О том, что у моего ребёнка синдром Прадера — Вилли, я узнала, лишь когда ему исполнилось шесть месяцев. И это стало одновременно и шоком, и облегчением. Теперь мы знали, с чем имеем дело и что делать дальше.
Как только анализы всё подтвердили, мы сели на диету. Потому что иначе нельзя. Главная проблема синдрома Прадера — Вилли — это гиперфагия, отсутствие чувства насыщения. Люди с таким заболеванием постоянно хотят есть, поэтому ожирение — основная проблема.

Фото из личного архива Марины Амулиной
Самая большая опасность — это смерть от удушья из-за ожирения и задержек дыхания во сне. Важно строго контролировать питание ребёнка. Исключается все сладкое вплоть до фруктов. Запрещено есть мучное. Интервалы в питании должны быть по четыре с половиной часа. Но важно, что у каждого ребёнка это индивидуально, поэтому нужна консультация грамотного врача. Мы регулярно делаем УЗИ, смотрим, как усваивается еда. Если через три часа в желудке она еще есть, то кормить ни в коем случае нельзя. Важно уменьшать калории, несмотря на то что это растущий организм. Каждые полтора месяца мы лежим на реабилитации. Между этим дома проводим медикаментозное лечение, делаем массажи.
Ни в одной стране нет волшебной таблетки от поломки в 15-й хромосоме, которая может вылечить такого ребёнка
Всё лечение симптоматическое. Ребёнок рождается, как правило, с недоразвитыми органами, потому что у него была внутриутробная гипотония и недостаток кислорода — при генетических нарушениях уже во время беременности начинаются проблемы. Опытные врачи могут увидеть, что с ребёнком что-то не так. Есть характерные признаки.
Знание диагноза помогает нам, родителям, знать, чего ожидать, какие меры необходимо принимать. И понимать, чего делать категорически нельзя.
Нам повезло, мы живем в Москве и постоянную реабилитацию проходим бесплатно. Детям из регионов, как правило, надо получать квоты на такое лечение, к каждому осведомленному о заболевании врачу приходится ездить из другого города, оплачивать билеты и жилье. Это дорого.
Лечение, которое нам доступно бесплатно, хорошего уровня. Всё, что моему ребёнку жизненно необходимо, ему дают. Конечно, есть реабилитации совсем другого уровня, намного лучше, но они очень дорогие. Цена варьируется от 250 до 700 тысяч рублей за один курс. А делать их нужно регулярно. В нашем случае это каждые полтора-два месяца. Также большинству деток с этим синдромом необходимы различные коррекционные операции.
Сейчас Роберту 2 года и 3 месяца. Несмотря на то что у него сильная задержка в развитии, динамика у нас колоссальная. Он стал более самостоятельным, многому научился. Каждый новый звук, каждое новое движение — это настоящий праздник, огромная победа. Сегодня он умеет то, что уже под силу ребёнку в 9–10 месяцев.
Вообще, мы стараемся не акцентировать внимание на болезни. Я сразу исключила из головы историю, что у меня ребёнок с проблемами. Просто воспринимаю это как ситуацию, с которой нужно работать. Никто не относится к Роберту как к особенному или неполноценному. Он растет в огромной любви. У него есть старший брат, который тоже еще маленький. Они сильно привязаны друг к другу, поддерживают в трудные моменты. Если бы хоть один член нашей семьи постоянно думал о болезни, то, наверное, мы бы все сошли с ума.

Фото из личного архива Марины Елян
Малая информированность врачей и родителей о СПВ приводит к тому, что многие узнают о заболевании ребёнка, когда уже присутствует сильное ожирение. Нам повезло, узнали достаточно рано. Но мы очень хотим, чтобы в мире и России это заболевание можно было диагностировать сразу при рождении, и еще лучше — при скрининге беременных. Это и сейчас возможно, но в список обязательных скринингов синдром не входит.
О создании благотворительного фонда помощи людям с синдромом Прадера — Вилли я задумалась сразу же, как узнала о диагнозе сына. Ведь нам самим некуда было податься в тот момент, подобных организаций не было, да даже научные статьи — почти все только иностранные. Я уже тогда понимала, как могу помочь родителям, которые находятся в такой же ситуации.
У меня есть огромная поддержка в виде семьи и мой опыт спокойно принимать проблемы и работать с ними конструктивно. Но у большинства родителей процесс принятия факта неизлечимого заболевания у ребенка проходит намного тяжелее. Многие впадают в депрессию, не понимают, как жить дальше, куда бежать со своими проблемами.
Немалым фактором становится еще и то, что русскоязычной информации об этом синдроме почти нет, нет сообществ для таких родителей
Я всегда много работала (у нас семейный бизнес), и это позволило избежать депрессивных состояний. Работа действительно хорошо вытянула меня в реальность, где нельзя раскисать. У меня есть старший сын, который также нуждается в веселой, счастливой маме, поэтому я быстро взяла себя в руки. Судьба нас свела с другими прекрасными мамами, чьи дети тоже родились с этим синдромом. Так что я очень счастлива, что быстро нашлись единомышленники и вместе мы создали фонд!
«Врач-генетик перинатального центра предположила спинально-мышечную дистрофию»
Марина Вербивская, мама ребёнка с СПВ, учредитель и президент фонда
Синдром Прадера — Вилли — редкое генетическое заболевание, вызванное поломкой в 15-й хромосоме. В большинстве случаев нарушение происходит случайно, никто из родителей не виноват. «Так сложились звезды», — услышала я однажды на приеме врача.
Когда я забеременела, мы с мужем сразу верили, что у нас будет девочка. Мое самочувствие и анализы — все было в норме. Ближе к 30-й неделе появилось многоводие, а с ним и тяжесть, дискомфорт. Я чувствовала, что малышка мало двигается внутри, но количество шевелений плода умещалось в норму, установленную врачами. Но ещё тогда нам с мужем стало понятно, что нас ожидает какая-то особенная история.
Я стала искать специалистов в области патологии беременности в лучших перинатальных центрах Москвы, и лишь в одном из них результат УЗИ показал все нарушения, соответствующие хромосомной аномалии плода и нарушению маточно-плацентарного кровотока. Но врач, которая могла тогда принять решение о тактике дальнейшего ведения беременности, доктор медицинских наук, профессор, не обратила на это внимания.
Рожать я отправилась в срок, предварительно наблюдаясь в стационарном отделении патологии беременности четыре дня, каждый из которых мой лечащий врач пропускал главный симптом, который наблюдается у каждой мамы ребенка с синдромом Прадера — Вилли при обязательном исследовании КТГ (кардиотокографии) плода. Роды начались естественные, но закончились экстренным кесаревым сечением. И это не редкая ошибка врачей, виной чему, конечно же, неосведомленность.
После операции меня отправили в отделение интенсивной терапии, а ребенка — с неплохими показателями, но небольшим весом и ростом — в детское отделение. Через несколько часов после рождения, когда материнские гормоны перестали работать, малышка сильно ослабла. Не плакала, не ела, практически не реагировала на сторонние раздражители. Ее поместили в отделение интенсивной терапии, и мы виделись с ней только два раза в день.
Она лежала в кувезе, я могла лишь трогать её руку, а она в ответ слегка сжимала мой палец и иногда открывала глаза
Почти сразу врач-генетик перинатального центра предположила СМА (спинально-мышечную дистрофию). Это один из самых страшных диагнозов, с которым большинство родителей детей с синдромом Прадера — Вилли живут первые пару месяцев жизни, пока не получат результаты, исключающие его. Некоторые опытные генетики уже могут найти отличия в синдромах, но остальным не хватает осведомленности.

Фото: Юлия Кондратова
После недели пребывания в перинатальном центре мы с Кристиной ещё месяц лежали в отделении педиатрии и патологии новорожденных. Весь месяц я плакала. Она была слишком слаба, и никто не понимал, что с ней. Когда я совершенно устала и потеряла надежду на улучшения, я решила, что хочу, чтобы Кристина была дома, где ее окружат братья и сестры, бабушки и дедушки и будут любить, несмотря ни на что.
За день до выписки лечащий врач организовала консультацию их генетика. Он и предположил синдром Прадера — Вилли. Я не верила в диагноз и не сразу заглянула в карту. А вечером все-таки отыскала медсестру и попросила прочитать название синдрома, анализ на который дочери рекомендовали сдать. Ввела его в поисковик и проплакала еще сутки. Но это были мои последние слезы. Мы вернулись домой и стали придумывать, как жить дальше с синдромом Прадера — Вилли.
У этого синдрома много проявлений и признаков, затрагивающих почти все системы организма, но они не обязательно проявятся все одновременно, да и их тяжесть варьируется от лёгкой до тяжелой. Самыми частыми и значимыми симптомами считаются сильнейшая гипотония (мышечная слабость) в младенчестве и сильнейшая гиперфагия (неконтролируемое переедание), приводящая к морбидному ожирению. Эти симптомы вызваны нарушениями эндокринной системы. Единственным эффективным средством лечения сегодня считается рекомбинантный гормон роста, который здорово справляется с гипотонией, но не сводит на нет чувство голода. Поэтому изобретения лекарства сегодня очень ждут во всем мире.
В России не ведется регистр пациентов с этим синдромом. За два года в закрытых родительских сообществах я встретила, наверное, около 300 человек
Но всем понятно, исходя из данных других стран о частоте возникновения синдрома (1 из 15–30 тысяч живых новорожденных), что в России таких людей тысячи.
Фото: Юлия Кондратова
Наш фонд помощи людям с синдромом Прадера — Вилли только начал свою работу. В рамках месяца осведомленности мы разместили в соцсетях 15 фактов о заболевании под хэштегом #знаюпроспв. Мы запустили сайт, подключаем платежные системы и ищем источники финансирования для реализации в том числе и программ оказания адресной помощи.
Наша главная цель — максимально расширить осведомленность о синдроме
Так как многое в жизни людей с синдромом Прадера — Вилли зависит от нескольких видов коррекций — гормональной, педагогической, поведенческой, коррекции физического развития — и строгой диеты.
Наша задача сводится к созданию информационной базы для каждого из участвующих в жизни людей с СПВ. Мы планируем создать как площадку для общения и осведомленности медиков любого уровня, от поликлиник до научных центров, так и информационные материалы для педагогов и воспитателей. А еще нам важно помочь семьям строить взаимоотношения в новой реальности, ведь, как бы ни сложилось течение заболевания, многое в их жизни теперь будет по-другому.
Быт, распределение времени, общение с родственниками и друзьями, традиции празднования дней рождений и других праздников. И будущее. Достаточно болезненная тема для каждого родителя — это взрослая жизнь наших детей. И наша задача — сопровождать людей с синдромом Прадера — Вилли на каждом этапе их жизни.
Родители — это люди, которые максимально заинтересованы в осведомленности о заболевании ребенка. Обычный врач в поликлинике может ни разу в жизни не столкнуться с таким диагнозом. Сегодня известно около 7000 редких орфанных заболеваний. При этом каждую неделю в медицинской литературе описывается в среднем пять новых.

Фото: Мария Трумпокаис
Например, только в 2017 году было открыто заболевание «синдром Шааф-Янга» (SYS), вызванное мутацией только одного из генов в участке синдрома Прадера — Вилли. Сейчас активно изучается связь между синдромами, и не исключено, что результатом этих исследований станет понимание нарушений пищевого поведения в СПВ, но об этих фактах в России почти неизвестно.
Становится очевидным, что именно открытие пациентской организации в России может во многом изменить жизни наших детей, наших семей, наших врачей, наших педагогов.
Сейчас Кристинке два года. Она даже умеет ходить, что достаточно круто и не обязательно для наших детей в этом возрасте. Все дети сильно отличаются друг от друга, у каждого свой индивидуальный план развития. Главное — это любовь и общение. И, конечно, много информации о будущих возможных сложностях, чтобы уже сейчас не допускать возможности осложнений здоровья.
Мы не знаем, как дальше будет развиваться Кристинка. Пойдет ли она в обычную школу или будет учиться в коррекционной; возможно, даже на надомном обучении. Будет ли она работать и жить самостоятельно, когда вырастет, или же в течение всей жизни ей будет необходим специальный уход. Будет ли она радовать нас своим здоровьем и заниматься спортом и живописью, или ей потребуется ряд операций, чтобы поддерживать жизнедеятельность. Мы не знаем. И каждая мысль о возможных сложностях завтра заставляет нас еще больше наслаждаться дочерью сегодня.
Источник
Что такое синдром Прадера — Вилли?
Синдром Прадера — Вилли (СПВ) — это генетическое заболевание, которое встречается примерно у одного из каждых 15 000 новорожденных.
Синдром Прадера — Вилли поражает мужчин и женщин с одинаковой частотой и затрагивает все расы и этнические группы. СПВ признана наиболее распространенной генетической причиной опасного для жизни детского ожирения.
Данный синдром был впервые описан швейцарскими врачами Андреа Прадером, Алексисом Лабхартом и Генрихом Вилли в 1956 году на основании клинических характеристик девяти обследованных детей.
Общие характеристики, определенные в первоначальном отчете, включали маленькие руки и ноги, аномальный рост и состав тела (маленький рост, очень низкая мышечная масса тела и раннее детское ожирение), мышечная гипотония (низкий мышечный тонус (слабость мышц)) при рождении, ненасытный голод, сильное ожирение и умственная отсталость.
Это видео даст краткое понимание проявлений болезни:
Синдром Прадера — Вилли является результатом аномалии хромосомы 15, и постановка точного диагноза сегодня основывается на генетических анализах.
Каковы симптомы синдрома Прадера — Вилли?
Симптомы синдрома Прадера — Вилли, вероятно, связаны с дисфункцией части мозга, называемой гипоталамус. Гипоталамус — это небольшой эндокринный орган у основания мозга, который играет важную роль во многих функциях организма, включая регулирование голода и сытости, температуры тела, боли, баланса сна и бодрствования, водного баланса, эмоций и фертильности.
Хотя считается, что дисфункция гипоталамуса приводит к появлению симптомов синдрома Прадера — Вилли, еще не ясно, как генетическая аномалия вызывает дисфункцию гипоталамуса.
У людей с данным синдромом симптомы со временем меняются. В целом, есть два основных этапа симптомов, связанных с СПВ:
Ранний период жизни
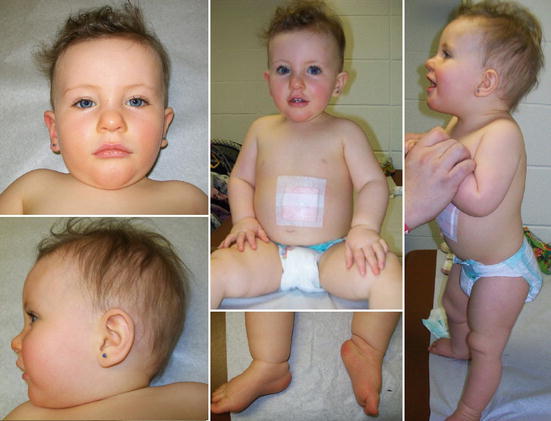
Младенцы с СПВ являются гипотоническими или «гибкими», с очень низким мышечным тонусом. Для них типичен слабый крик и плохой сосательный рефлекс. Детей с синдромом Прадера — Вилли обычно не могут кормить грудью и они часто нуждаются в искусственном вскармливании.
У детей могут быть проблемы с ростом и развитием, если трудности с кормлением не подвергаются тщательному мониторингу и лечению. По мере взросления этих детей сила и мышечный тонус в целом улучшаются. Моторные функции развиты, но, как правило, выполняются с задержкой.
Малыши, как правило, вступают в период, когда они могут легко начать набирать вес до того, как проявят повышенный интерес к еде.
Детство и взросление
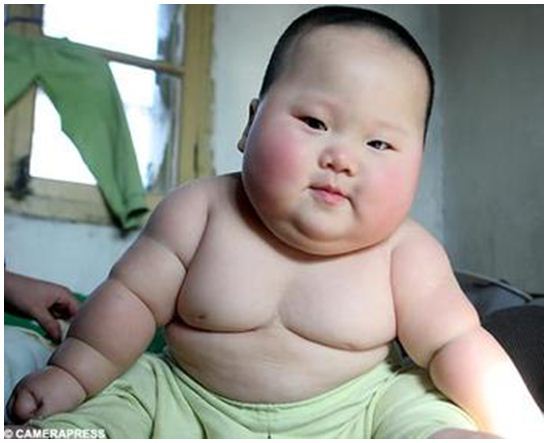
Нерегулируемый аппетит и легкое увеличение веса (см. фото) характеризуют более поздние стадии синдрома Прадера — Вилли.
Эти признаки чаще всего начинаются в возрасте от 3 до 8 лет, но различаются по началу и интенсивности.
Люди с синдромом Прадера — Виллии испытывают недостаток в нормальных подсказках голода и сытости.
Как правило, они не могут контролировать потребление пищи и переедают, если их не контролировать.
Поведение в голоде очень распространено. Кроме того, скорость метаболизма у людей с данным синдромом ниже, чем обычно. Без лечения эта комбинация проблем приводит к патологическому ожирению и его многочисленным осложнениям.
Помимо ожирения, у ребенка с синдромом Прадера — Вилли может быть множество других симптомов. Дети с IQ от низкого нормального до умеренной умственной отсталости обычно имеют когнитивные проблемы. Те, у кого нормальный IQ, обычно имеют проблемы с обучаемостью.
Другие проблемы и признаки могут включать:
- дефицит гормона роста/низкий рост;
- маленькие руки и ноги;
- сколиоз;
- нарушения сна с чрезмерной дневной сонливостью;
- высокий болевой порог;
- апраксия/диспраксия речи и бесплодие.
Поведенческие трудности могут включать в себя обсессивно-компульсивные симптомы, проблемы с кожей и трудности с контролем эмоций. Взрослые с синдромом Прадера— Вилли подвергаются повышенному риску психических заболеваний. СПВ является расстройством широкого спектра, и симптомы варьируют по степени тяжести и частоты встречаемости среди людей.
Хромосомы и гены: основы
Чтобы понять генетику СПВ, необходимо иметь общее представление о хромосомах и генах.
Хромосомы представляют собой крошечные структуры, которые присутствуют почти в каждой клетке нашего тела. Это пакеты генов, которые мы наследуем от наших родителей. Гены содержат все подробные инструкции, необходимые нашему телу для роста, развития и функционирования — нашу ДНК.
Определенные гены направляют наши клетки на производство белков, ферментов и других необходимых веществ. Каждый из наших многочисленных генов находится на определенной хромосоме. Большинство клеток нашего тела содержат 46 хромосом — 23 унаследованы от нашей матери и 23 от нашего отца. (Яйца и сперматозоиды обычно содержат всего 23 хромосомы, потому что это те клетки объединяются в зачатии и обеспечивают ребенку нужное количество хромосом.)
22 пары хромосом помечены числом, основанным на их размере (хромосома 1 — самая большая пара, а хромосома 22 — почти наименьшая), и 2 хромосомы в каждой пронумерованной паре содержат одинаковые гены (один набор у матери и одну от отца). Изменения, которые вызывают синдром Прадера — Вилли, происходят в паре, известной как хромосома 15. 23-я пара хромосом обозначена как пара половых хромосом. Эта пара определяет пол ребенка: XX для девочки, XY для мальчика.
Изменения или ошибки в генах и хромосомах распространены при образовании яйцеклеток и сперматозоидов. Некоторые из этих генетических изменений не будут иметь эффекта, когда ребенок зачат; некоторые вызовут выкидыш; а некоторые спровоцируют, например, синдром Прадера — Вилли, вызвав значительные различия в том, как ребенок развивается и функционирует.
В то время как многие генетические нарушения вызваны изменением одного гена и могут передаваться от родителя к ребенку, СПВ куда более сложен.
Что вызывает синдром Прадера — Вилли (причины)?
Синдром Прадера — Вилли возникает в результате отсутствия активного генетического материала в определенной области хромосомы 15 (15q11-q13). Обычно люди наследуют одну копию хромосомы 15 от своей матери и одну от своего отца. Гены вызывающие СПВ активны только в хромосоме, полученной от отца.
При синдроме Прадера — Вилли генетический дефект, вызывающий неактивность хромосомы 15 от отца (отцовская хромосома 15), может возникнуть по трем следующим причинам:
- СПВ по исключению: чаще всего часть хромосомы 15, которая была унаследована от отца, отсутствует или удалена в этой критической области. Эта небольшая делеция встречается примерно в 70% случаев и обычно не выявляется при обычном генетическом анализе, таком как амниоцентез.
- СПВ по однородительской дисомии: еще около 30% случаев происходят, когда человек наследует две хромосомы 15 от своей матери и ни один от своего отца. Это тип наследование называется однородительской дисомией (UPD).
- СПВ по импрессии мутации: наконец, в очень небольшом проценте случаев (1-3%) небольшая генетическая мутация при синдроме Прадера — Вилли приводит к тому, что генетический материал отцовской хромосомы 15 хоть и присутствует, но неактивен.
Как эти генетические дефекты вызывают симптомы, наблюдаемые при синдроме Прадера — Вилли?
Хромосомы 15 одни из самых сложных областей генома человека. Хотя были достигнуты значительные успехи в понимании и характеристике генетических изменений, связанных с синдромом Прадера — Вилли, точный механизм, с помощью которого отсутствие функционального генетического материала приводит к симптомам, связанным с СПВ, не понятны.
Ученые активно изучают нормальную роль генетических последовательностей в области СПВ и то, как их потеря влияет на гипоталамус и другие системы организма.
Существуют ли различия в степени тяжести СПВ в зависимости от генетического подтипа?
Могут быть некоторые тонкие различия в признаках СПВ на основе генетического подтипа: например, лица с делециями могут быть светлокожими со светлыми волосами по сравнению с другими членами семьи и быть более восприимчивыми к припадкам и судорогам; а те, у кого есть проблемы с СПВ по однородительской дисомии, могут быть подвержены более высокому риску психических заболеваний в молодом подростковом возрасте.
В целом, однако, существует значительное совпадение между различными генетическими подтипами. Вероятно, что тысячи генов за пределами области СПВ, которые демонстрируют нормальные различия между индивидуумами, также вносят значительный вклад в вариабельность симптомов синдрома Прадера — Вилли между теми, у кого есть расстройство.
Как диагностируется СПВ?
Данный синдром диагностируется с помощью анализа крови, который выявляет генетические аномалии, специфичные для синдрома Прадера — Вилли — так называемый «анализ метилирования ДНК». Метод FISH (флуоресцентная гибридизация in-situ) идентифицирует синдром путем делеции, но не диагностирует другие формы СПВ.
Анализ на метилирование ДНК идентифицирует все типы этого синдрома и является предпочтительным методом исследования для диагностики. Если анализ на метилирование проводится первым, может потребоваться дополнительное исследование, чтобы определить, вызван ли СПВ отцовской делецией, UPD или импринтинговой мутацией.
В случаях, когда существует подозрение на наличие импринтинговой мутации, кровь также может быть взята у родителей.
Каковы риски синдрома Прадера — Вилли в будущем?
СПВ по исключению и UPD являются случайными явлениями и, как правило, не связаны с повышенным риском рецидива в будущих беременностях. В случае импринтинг-мутации синдром Прадера — Вилли может в семье повториться. Семьи с опасениями по поводу их риска СПВ должны поговорить с генетическим консультантом.
Есть ли лекарство от синдрома Прадера — Вилли?
В настоящее время нет никакого лечения от синдрома Прадера — Вилли, и большинство исследований на сегодняшний день было направлено на лечение определенных симптомов данного синдрома.
Для многих людей, затронутых этим расстройством, устранение некоторых из самых сложных аспектов синдрома, таких как ненасытный аппетит и ожирение, будет представлять собой значительное улучшение качества жизни и способности жить независимо.
Источник