Дети с синдромом прадера вилли фото

Что такое синдром Прадера — Вилли?
Синдром Прадера — Вилли (СПВ) — это генетическое заболевание, которое встречается примерно у одного из каждых 15 000 новорожденных.
Синдром Прадера — Вилли поражает мужчин и женщин с одинаковой частотой и затрагивает все расы и этнические группы. СПВ признана наиболее распространенной генетической причиной опасного для жизни детского ожирения.
Данный синдром был впервые описан швейцарскими врачами Андреа Прадером, Алексисом Лабхартом и Генрихом Вилли в 1956 году на основании клинических характеристик девяти обследованных детей.
Общие характеристики, определенные в первоначальном отчете, включали маленькие руки и ноги, аномальный рост и состав тела (маленький рост, очень низкая мышечная масса тела и раннее детское ожирение), мышечная гипотония (низкий мышечный тонус (слабость мышц)) при рождении, ненасытный голод, сильное ожирение и умственная отсталость.
Это видео даст краткое понимание проявлений болезни:
Синдром Прадера — Вилли является результатом аномалии хромосомы 15, и постановка точного диагноза сегодня основывается на генетических анализах.
Каковы симптомы синдрома Прадера — Вилли?
Симптомы синдрома Прадера — Вилли, вероятно, связаны с дисфункцией части мозга, называемой гипоталамус. Гипоталамус — это небольшой эндокринный орган у основания мозга, который играет важную роль во многих функциях организма, включая регулирование голода и сытости, температуры тела, боли, баланса сна и бодрствования, водного баланса, эмоций и фертильности.
Хотя считается, что дисфункция гипоталамуса приводит к появлению симптомов синдрома Прадера — Вилли, еще не ясно, как генетическая аномалия вызывает дисфункцию гипоталамуса.
У людей с данным синдромом симптомы со временем меняются. В целом, есть два основных этапа симптомов, связанных с СПВ:
Ранний период жизни
Младенцы с СПВ являются гипотоническими или «гибкими», с очень низким мышечным тонусом. Для них типичен слабый крик и плохой сосательный рефлекс. Детей с синдромом Прадера — Вилли обычно не могут кормить грудью и они часто нуждаются в искусственном вскармливании.
У детей могут быть проблемы с ростом и развитием, если трудности с кормлением не подвергаются тщательному мониторингу и лечению. По мере взросления этих детей сила и мышечный тонус в целом улучшаются. Моторные функции развиты, но, как правило, выполняются с задержкой.
Малыши, как правило, вступают в период, когда они могут легко начать набирать вес до того, как проявят повышенный интерес к еде.
Детство и взросление
Нерегулируемый аппетит и легкое увеличение веса (см. фото) характеризуют более поздние стадии синдрома Прадера — Вилли.
Эти признаки чаще всего начинаются в возрасте от 3 до 8 лет, но различаются по началу и интенсивности.
Люди с синдромом Прадера — Виллии испытывают недостаток в нормальных подсказках голода и сытости.
Как правило, они не могут контролировать потребление пищи и переедают, если их не контролировать.
Поведение в голоде очень распространено. Кроме того, скорость метаболизма у людей с данным синдромом ниже, чем обычно. Без лечения эта комбинация проблем приводит к патологическому ожирению и его многочисленным осложнениям.
Помимо ожирения, у ребенка с синдромом Прадера — Вилли может быть множество других симптомов. Дети с IQ от низкого нормального до умеренной умственной отсталости обычно имеют когнитивные проблемы. Те, у кого нормальный IQ, обычно имеют проблемы с обучаемостью.
Другие проблемы и признаки могут включать:
- дефицит гормона роста/низкий рост;
- маленькие руки и ноги;
- сколиоз;
- нарушения сна с чрезмерной дневной сонливостью;
- высокий болевой порог;
- апраксия/диспраксия речи и бесплодие.
Поведенческие трудности могут включать в себя обсессивно-компульсивные симптомы, проблемы с кожей и трудности с контролем эмоций. Взрослые с синдромом Прадера— Вилли подвергаются повышенному риску психических заболеваний. СПВ является расстройством широкого спектра, и симптомы варьируют по степени тяжести и частоты встречаемости среди людей.
Хромосомы и гены: основы
Чтобы понять генетику СПВ, необходимо иметь общее представление о хромосомах и генах.
Хромосомы представляют собой крошечные структуры, которые присутствуют почти в каждой клетке нашего тела. Это пакеты генов, которые мы наследуем от наших родителей. Гены содержат все подробные инструкции, необходимые нашему телу для роста, развития и функционирования — нашу ДНК.
Определенные гены направляют наши клетки на производство белков, ферментов и других необходимых веществ. Каждый из наших многочисленных генов находится на определенной хромосоме. Большинство клеток нашего тела содержат 46 хромосом — 23 унаследованы от нашей матери и 23 от нашего отца. (Яйца и сперматозоиды обычно содержат всего 23 хромосомы, потому что это те клетки объединяются в зачатии и обеспечивают ребенку нужное количество хромосом.)
22 пары хромосом помечены числом, основанным на их размере (хромосома 1 — самая большая пара, а хромосома 22 — почти наименьшая), и 2 хромосомы в каждой пронумерованной паре содержат одинаковые гены (один набор у матери и одну от отца). Изменения, которые вызывают синдром Прадера — Вилли, происходят в паре, известной как хромосома 15. 23-я пара хромосом обозначена как пара половых хромосом. Эта пара определяет пол ребенка: XX для девочки, XY для мальчика.
Изменения или ошибки в генах и хромосомах распространены при образовании яйцеклеток и сперматозоидов. Некоторые из этих генетических изменений не будут иметь эффекта, когда ребенок зачат; некоторые вызовут выкидыш; а некоторые спровоцируют, например, синдром Прадера — Вилли, вызвав значительные различия в том, как ребенок развивается и функционирует.
В то время как многие генетические нарушения вызваны изменением одного гена и могут передаваться от родителя к ребенку, СПВ куда более сложен.
Что вызывает синдром Прадера — Вилли (причины)?
Синдром Прадера — Вилли возникает в результате отсутствия активного генетического материала в определенной области хромосомы 15 (15q11-q13). Обычно люди наследуют одну копию хромосомы 15 от своей матери и одну от своего отца. Гены вызывающие СПВ активны только в хромосоме, полученной от отца.
При синдроме Прадера — Вилли генетический дефект, вызывающий неактивность хромосомы 15 от отца (отцовская хромосома 15), может возникнуть по трем следующим причинам:
- СПВ по исключению: чаще всего часть хромосомы 15, которая была унаследована от отца, отсутствует или удалена в этой критической области. Эта небольшая делеция встречается примерно в 70% случаев и обычно не выявляется при обычном генетическом анализе, таком как амниоцентез.
- СПВ по однородительской дисомии: еще около 30% случаев происходят, когда человек наследует две хромосомы 15 от своей матери и ни один от своего отца. Это тип наследование называется однородительской дисомией (UPD).
- СПВ по импрессии мутации: наконец, в очень небольшом проценте случаев (1-3%) небольшая генетическая мутация при синдроме Прадера — Вилли приводит к тому, что генетический материал отцовской хромосомы 15 хоть и присутствует, но неактивен.
Как эти генетические дефекты вызывают симптомы, наблюдаемые при синдроме Прадера — Вилли?
Хромосомы 15 одни из самых сложных областей генома человека. Хотя были достигнуты значительные успехи в понимании и характеристике генетических изменений, связанных с синдромом Прадера — Вилли, точный механизм, с помощью которого отсутствие функционального генетического материала приводит к симптомам, связанным с СПВ, не понятны.
Ученые активно изучают нормальную роль генетических последовательностей в области СПВ и то, как их потеря влияет на гипоталамус и другие системы организма.
Существуют ли различия в степени тяжести СПВ в зависимости от генетического подтипа?
Могут быть некоторые тонкие различия в признаках СПВ на основе генетического подтипа: например, лица с делециями могут быть светлокожими со светлыми волосами по сравнению с другими членами семьи и быть более восприимчивыми к припадкам и судорогам; а те, у кого есть проблемы с СПВ по однородительской дисомии, могут быть подвержены более высокому риску психических заболеваний в молодом подростковом возрасте.
В целом, однако, существует значительное совпадение между различными генетическими подтипами. Вероятно, что тысячи генов за пределами области СПВ, которые демонстрируют нормальные различия между индивидуумами, также вносят значительный вклад в вариабельность симптомов синдрома Прадера — Вилли между теми, у кого есть расстройство.
Как диагностируется СПВ?
Данный синдром диагностируется с помощью анализа крови, который выявляет генетические аномалии, специфичные для синдрома Прадера — Вилли — так называемый «анализ метилирования ДНК». Метод FISH (флуоресцентная гибридизация in-situ) идентифицирует синдром путем делеции, но не диагностирует другие формы СПВ.
Анализ на метилирование ДНК идентифицирует все типы этого синдрома и является предпочтительным методом исследования для диагностики. Если анализ на метилирование проводится первым, может потребоваться дополнительное исследование, чтобы определить, вызван ли СПВ отцовской делецией, UPD или импринтинговой мутацией.
В случаях, когда существует подозрение на наличие импринтинговой мутации, кровь также может быть взята у родителей.
Каковы риски синдрома Прадера — Вилли в будущем?
СПВ по исключению и UPD являются случайными явлениями и, как правило, не связаны с повышенным риском рецидива в будущих беременностях. В случае импринтинг-мутации синдром Прадера — Вилли может в семье повториться. Семьи с опасениями по поводу их риска СПВ должны поговорить с генетическим консультантом.
Есть ли лекарство от синдрома Прадера — Вилли?
В настоящее время нет никакого лечения от синдрома Прадера — Вилли, и большинство исследований на сегодняшний день было направлено на лечение определенных симптомов данного синдрома.
Для многих людей, затронутых этим расстройством, устранение некоторых из самых сложных аспектов синдрома, таких как ненасытный аппетит и ожирение, будет представлять собой значительное улучшение качества жизни и способности жить независимо.
Источник
Ожирение в современном цивилизованном обществе приняло характер эпидемии. Всё больше и больше людей страдают от недостатка физической нагрузки и избыточного веса. Среди больных с ожирением отдельную группу составляют дети и подростки. Причины развития заболевания в этом случае заключаются не только в малоподвижном образе жизни и несбалансированном питании, но и в генетических аномалиях. К подобным патологиям относится синдром Прадера — Вилли.
Наследственное ожирение: определение и вероятность развития
Вся информация о внешности человека, характере обменных процессов в организме заложена в молекуле дезоксирибонуклеиновой кислоты (ДНК). Участки ДНК, кодирующие определённые признаки (цвет волос, глаз, кожи), носят название генов. Весь наследственный материал человека сгруппирован в сорок шесть хромосом, отлично видимых в ядре клетки через микроскоп. Половину из них будущий ребёнок получает от отца, другую — от матери. Хромосомы первоначально содержатся в половых клетках — яйцеклетке и сперматозоиде.
Хромосомный набор человека содержит всю информацию о строении и функционировании организма
Любое повреждение молекулы ДНК различной протяжённости (ген, участок хромосомы) с большой вероятностью скажется или на внешних чертах человека, или на характере обмена веществ. Синдром Прадера — Вилли — медицинский термин, описывающий характерные изменения внешнего вида больного в сочетании с ожирением наследственной природы и особенностями обмена веществ.
Синдром Прадера — Вилли — совокупность ожирения, изменённых черт лица и пропорций тела наследственной природы
Впервые синдром был описан учёными Андреасом Прадером и Генрихом Вилли в 1956 году. Распространённость патологии составляет один случай на десять — двадцать тысяч новорождённых. Заболевание одинаково часто встречается среди мальчиков и девочек. В литературе описаны семейные случаи синдрома Прадера — Вилли.
Наследственность как основная причина заболевания
Основная причина развития синдрома Прадера — Вилли — нарушение в структуре генов, расположенных в пятнадцатой хромосоме. К формированию болезни приводят два типа дефекта:
- потеря большого участка молекулы ДНК (микроделеция) отцовского происхождения;
- наследование обеих пятнадцатых хромосом от матери (изодисомия).
Синдром Прадера — Вилли развивается вслествие генетических дефектов пятнадцатой хромосомы
Около двух третей случаев патологии обусловлены микроделецией, остальные — материнской изодисомией.
Существует генетический противоположный дефект: микроделеция материнского происхождения или отцовская дисомия. Обе причины приводят к развитию наследственной патологии под названием синдром Ангельмана. Клиническая картина при этом заболевании характеризуется замедлением физического и интеллектуального развития, нарушением координации движений, эпилептическими судорожными приступами.
До развития молекулярной генетики было принято считать, что наследование обеих пар хромосом от одного родителя невозможно. Однако с появлением современных методов анализа был не только доказан этот факт. В процессе проводимых генетических исследований было опровергнуто утверждение о равноценном влиянии отцовской и материнской наследственной информации на внешние черты и характер обмена веществ ребёнка.
Клинические аспекты генетических заболеваний — видео
Особенности патологии
Основным следствием генетических аномалий при синдроме Прадера — Вилли является необычный обмен жиров в организме, приводящий к десятикратному превалированию их отложения в подкожно-жировой клетчатке над расщеплением. Ещё одним важным механизмом развития заболевания является нарушение обмена половых гормонов, в результате которого репродуктивная система имеет множество аномалий анатомического строения.
Кроме того, при синдроме Прадера — Вилли значительно повышен риск образования злокачественных опухолей вследствие генетически запрограммированной слабой системы защиты молекулы ДНК от поломок.
Симптомы заболевания
Клиническая картина при синдроме Прадера — Вилли у детей разного возраста существенно различается.
Признаки синдрома Прадера — Вилли в различных возрастных группах — таблица
| Возрастные группы | Новорождённые | 12–18 месяцев | Дошкольники и подростки |
| Показатели | |||
| Физическое развитие |
| Выраженное снижение мышечного тонуса |
|
| Нервно-психическое развитие | Соответствует возрасту | Ослабление рефлексов | Задержка интеллектуального развития |
| Половое развитие | Соответствует возрасту, возможно отсутствие яичек в мошонке (крипторхизм) | Соответствует возрасту, возможно отсутствие яичек в мошонке (крипторхизм) | Недоразвитие половых органов, крипторхизм |
| Анатомические аномалии | Незначительно увеличены размеры черепа | Незначительно увеличены размеры черепа |
|
В период внутриутробного развития плод с синдромом Прадера — Вилли имеет незначительные анатомические особенности в виде узкого лицевого скелета.
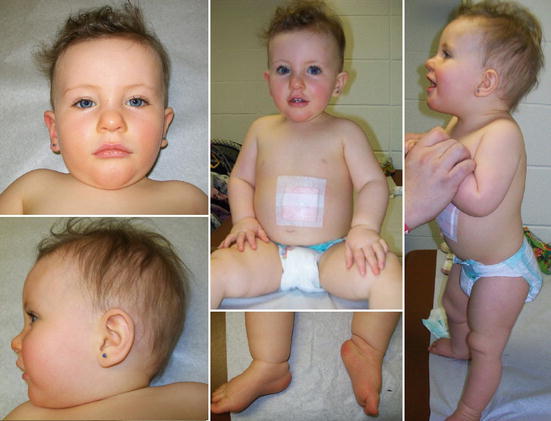
Внешний вид больных с синдромом Прадера — Вилли — фото
Методы диагностики
Для установления диагноза используются следующие методики:
- тщательный осмотр врача — обязательно с выяснением всех деталей заболевания, включая особенности периода беременности, родов и ранних этапов развития ребёнка;
- измерение роста и веса, длины сегментов конечностей — проводится с целью определения важнейших признаков болезни: избыточного веса, низкого роста, диспропорции конечностей;
Диспропорция конечностей — один из основных внешних признаков синдрома Прадера — Вилли
- неврологический осмотр — для грудного ребёнка позволяет выявить ослабление рефлексов и снижение мышечного тонуса. В более старшем возрасте осмотр преследует цель определить степень нарушения интеллекта;
- анализ крови на уровень гормонов — позволяет выявить нарушение функций яичек и яичников;
- биохимический анализ крови — позволяет выявить признаки сопутствующего сахарного диабета в виде повышенного уровня глюкозы;
- ультразвуковое исследование внутренних органов — позволяет выявить сопутствующие анатомические аномалии их строения;
- осмотр офтальмолога — позволяет выявить снижение остроты зрения;
- электронейромиография — позволяет графически отобразить прохождение нервного сигнала по мышечным волокнам;
- молекулярно-генетическое исследование — является золотым стандартом и позволяет точно выявить дефект строения пятнадцатой хромосомы.
Выявление генетического дефекта — золотой стандарт диагностики синдрома Прадера — Вилли
Дифференциальный диагноз проводится со следующими заболеваниями, при которых основными признаками являются ожирение, низкий мышечный тонус, задержка интеллектуального развития:
- синдромом Дауна;
- миопатией;
- спинальной амиотрофией;
- синдромом Лоуренса — Муна;
- синдромом Барде — Бидля;
- синдромом Альстрема;
- синдромом Опитца — Фриаса.
Наследственные синдромы — фотогалерея
Лечение наследственного ожирения
Лечение заболевания направлено на улучшение качества жизни больного, поскольку генетический дефект на современном этапе развития медицины исправить невозможно. Терапия при синдроме Прадера — Вилли комплексная, осуществляется коррекция питания и веса, мышечного тонуса, полового развития, а также исходных интеллектуальных способностей.
Медикаментозное лечение
Лекарственные препараты используются с целью достижения приемлемого уровня роста, а также правильного формирования половых органов в пубертатный период. Гормон соматотропин способствует росту мышц и скелета. Препарат используется до момента закрытия хрящевых зон длинных костей плеча, бедра, голени и предплечья.
Правильное половое развитие при синдроме Прадера — Вилли достигается назначением гормона гонадотропина. Препарат обеспечивает нормальный процесс формирования вторичных половых признаков.
Важно помнить, что только врач может решить, какова будет продолжительность курса лечения, назначить соответствующие препараты.
Для формирования вторичных половых признаков подросткам с синдромом Прадера — Вилли назначается Хорионический гонадотропин
Физиотерапия
Физиотерапевтические процедуры, используемые в раннем возрасте для коррекции мышечного тонуса:
- электростимуляция;
- электрофорез.
В основе этих методов лежит благотворное воздействие электрического тока различной формы и полярности. Массаж позволяет создать гармоничный тонус в различных группах мышц. Лечебная гимнастика — необходимое мероприятие при терапии больных с синдромом Прадера — Вилли. Наибольший эффект достигается при занятиях плаванием и аквааэробикой. Достаточный уровень физической активности жизненно необходим пациенту в борьбе с лишним весом.
Плавание — идеальный вид спорта для больного с синдромом Прадера — Вилли
Диета
Рациональное питание — основной способ коррекции особенного обмена жиров у больных. Прежде всего необходимо приучить ребёнка не переедать, поэтому родителям необходимо исключить свободный доступ к пище.
Рекомендуется к употреблению следующий набор продуктов:
- хлеб из цельных злаков;
- яркоокрашенные ягоды;
- свежевыжатые соки;
- кисломолочные продукты;
- морская рыба;
- морепродукты;
- зелёный чай;
- свежие овощи и фрукты;
- морская капуста.
Фотогалерея: продукты, рекомендуемые для включения в рацион
Продукты, которых необходимо избегать:
- сахар;
- газированные напитки;
- фастфуд;
- жирные сорта мяса и рыбы;
- белый хлеб;
- картофель;
- шоколад;
- кондитерские изделия с кремом;
- свежая выпечка.
Фотогалерея: продукты, от которых следует отказаться
Коррекция интеллектуального развития ребёнка
Адекватный уровень интеллекта — залог социализации больных с синдромом Прадера — Вилли. Развитие исходных способностей на занятиях с логопедом-дефектологом и педагогом позволяет детям быть принятыми в любой коллектив.
Развитие правильного произношения и образной речи очень важно для ребёнка с синдромом Прадера — Вилли
Осложнения и прогноз заболевания
Своевременная постановка диагноза и адекватная терапия позволяет больным с синдромом Прадера — Вилли достичь приемлемого уровня качества жизни. При неблагоприятном течении заболевания развиваются следующие осложнения:
- сахарный диабет;
- тяжёлые формы остановки дыхания во время сна (апноэ);
- выраженное искривление позвоночника;
- разрушение хрящевой ткани суставов под влиянием избыточного веса;
- сердечная недостаточность;
- злокачественные новообразования.
При соблюдении всех врачебных рекомендаций продолжительность жизни больных с синдромом Прадера — Вилли может превышать шестьдесят лет.
Профилактика
Единственным эффективным методом профилактики является дородовая генетическая диагностика с определением особенностей хромосомного набора клеток плода, полученных из околоплодных вод. В последующем проводится консультирование врачом-генетиком.
Генетическое консультирование — единственный эффективный метод профилактики синдрома Прадера — Вилли
Синдром Прадера — Вилли — серьёзное генетическое заболевание. Для сохранения должного качества жизни необходимо длительное упорное сотрудничество ребёнка, его родителей и медиков. Только дисциплинированное выполнение всех рекомендаций сделает пациента полноценным членом современного общества.
- Автор: Елена Тимофеева
- Распечатать
Имею высшее медицинское образование и опыт работы
Оцените статью:
Источник