Дети с синдромом миллера дикера

Синдром Миллера-Дикера: причины, диагностика, лечениеЭтиология и встречаемость синдрома Миллера-Дикера. Синдром Миллера-Дикера (MDS, MIM №247200) -синдром протяженной делеции, вызываемый гемизиготной делецией 17р13.3; механизм, лежащий в основе повторных делеции участка 17р13.3, еще не объяснен, но, возможно (подобно другим синдромам микроделеции), он включает рекомбинацию между повторяющейся ДНК-последовательностью с небольшим числом копий. Синдром Миллера-Дикера — редкое заболевание неопределенной встречаемости во всех популяциях. Патогенез синдрома Миллера-ДикераВ регионе делеции синдрома Миллера-Дикера в 17р13.3 картировано более 50 генов, но только ген LIS1 (MIM №601545) связан со специфической фенотипической характеристикой синдрома; гемизиготность по LIS1 вызывает лиссэнцефалию. LIS1 кодирует мозговую изоформу некаталитической бета-субъединицы фактора активации ацетилгидролазы тромбоцитов (PAFAH). PAFAH — ингибитор миграции нейронов и также связывает и стабилизирует микрофибриллы. Предварительные наблюдения указывают, что PAFAH может играть роль в реорганизации микрофибрилл, необходимой для миграции нейронов. Тем не менее изолированная гаплонедостаточность LIS1 не вызывает других дисморфических признаков, характерных для синдрома Миллера-Дикера. Мутации в гене LIS1 вызывают изолированную лиссэнцефалию (MIM №607432), т.е. лиссэнцефалию без других дисморфии. Поскольку все пациенты с синдромом Миллера-Дикера имеют дисморфические черты лица, этот дисморфизм должен вызываться гаплонедостаточностью одного или нескольких других генов в делеции.
Фенотип и развитие синдрома Миллера-ДикераСимптоматика синдрома Миллера-Дикера включает дисгенезию мозга, мышечную гипотонию, задержку развития и лицевые дисморфии. Дисгенезия мозга характеризуется лиссэнцефалией I (полная агирия) или II типа (широко распространенная агирия с несколькими бороздами на лобном или затылочном полюсе), церебральной корой с четырьмя вместо шести слоев, гетеротопией серого вещества и истончением белого вещества. У некоторых больных также отмечают пороки развития сердца и омфалоцеле. Дети с синдромом Миллера-Дикера плохо едят и растут. Способность улыбаться, краткий визуальный контакт и неспецифические двигательные реакции — единственные способности, приобретаемые большинством пациентов. Кроме умственной отсталости, пораженные обычно страдают от опистотонуса, спастичности и судорог. Почти все они умирают к 2 годам жизни. Особенности фенотипических проявлений синдрома Миллера-Дикера:
Лечение синдрома Миллера-ДикераЧерты лица больных и обнаружение на МРТ лиссэнцефалии часто позволяет предположить диагноз синдрома Миллера-Дикера. Тем не менее для подтверждения диагноза необходимо обнаружить делецию 17р13.3 при хромосомном анализе или FISH с LIS1-специфическим зондом. Приблизительно 60% пациентов имеют видимую делецию критического региона синдрома Миллера-Дикера. Синдром Миллера-Дикера неизлечим; следовательно, помощь направлена на коррекцию симптомов и паллиативный уход. Почти всем детям необходимо лекAPCтвенное лечение судорог. Большинство пациентов получают питание через назогастральный или гастростомический зонд из-за проблем вскармливания и повторных аспирации. Риски наследования синдрома Миллера-ДикераУ 80% пациентов бывает вновь возникшая микроделеция 17р13.3, а 20% наследуют делецию от одного из родителей, несущего сбалансированную хромосомную перестройку. Из-за частоты, с которой деления наследуется от родителя со сбалансированной транслокацией, анализ кариотипа и FISH на LIS1 следует выполнять у обоих родителей. Родитель со сбалансированной транслокацией, захватывающей 17р13.3, имеет приблизительно один шанс из четырех иметь аномального живорожденного ребенка (с синдромом Миллера-Дикера или с дупликацией dupl7p) и приблизительно один шанс из пяти — прерывания беременности. В отличие от этого, если пациент имеет синдром Миллера-Дикера в результате новой делеции, родители имеют низкий риск для повторения синдрома у последующих детей. Хотя порок развития мозга при синдроме Миллера-Дикера вызван неполной миграцией нейронов в церебральную кору во время 3-4 месяцев гестации, лиссэнцефалию не обнаруживают при МРТ или ультрасонографии плода до конца беременности. Для пренатальной диагностики синдрома Миллера-Дикера необходимо обнаружение делеции 17р13.3 в ворсинах хориона или амнио-цитах плода. Пример синдрома Миллера-Дикера. Б.Б., мальчик 5 дней жизни, родившийся на 38-й нед гестации, переведен в палату интенсивной терапии отделения новорожденных из-за выраженной гипотонии и затруднений вскармливания. Ребенок родился от неосложненной беременности; УЗИ плода на 14-й нед гестации и материнский сывороточный скрининг на 16-й нед гестации соответствовали норме. Мальчик родился от спонтанных влагалищных родов; оценка по шкале Апгар была 8 баллов на 1-й мин и 9 баллов на 5-й мин жизни. В семейном анамнезе ребенка генетические, неврологические или врожденные заболевания отсутствуют. При клиническом осмотре выявлены мышечная гипотония и слегка дисморфиче-ские черты лица, включая сужение битемпорального расстояния, вдавленную переносицу, небольшой нос с вывернутыми ноздрями и микрогнатию. В остальном данные обследования в норме. Его анализы на электролиты сыворотки крови, метаболический скрининг и врожденные инфекции оказались в пределах нормы. Ультразвуковое сканирование мозга показало гипоплазию мозолистого тела, легкое расширение желудочков мозга и сглаженную кору. После дополнительного совещания группа генетиков рекомендовала хромосомный анализ, флюоресцентный анализ (FISH) гена LIS1 (расположенный в регионе 17р13.3) и МРТ мозга. МРТ показала уплотнение коры мозга, полную агирию, многочисленные церебральные гетеротопии, гипоплазию мозолистого тела, нормальный мозжечок и ствол мозга. Хромосомный анализ с G-окраской оказался нормальным (46,XY), но FISH показал деле-цию LIS1 в одной из хромосом 17. На основе этих результатов генетик объяснил родителям, что у ребенка синдром Миллера-Дикера. Родители отказались от лечебных мероприятий, кроме необходимых для комфорта ребенка, и он умер в 2-месячном возрасте. — Также рекомендуем «Миоклонус-эпилепсия MERRF: причины, диагностика, лечение» Оглавление темы «Наследственные болезни»:
|
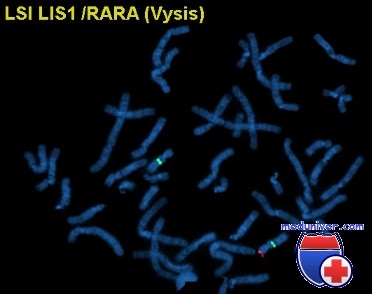
Источник
Синдром Миллера-Дикера — очень редкий синдром делеции 17p13.3. Этот синдром характеризуется лиссэнцефалией классического типа, отличительными чертами лица и другими врожденными пороками, которые встречаются только у некоторых лиц с этим синдромом.
Синдром Миллера-Дикера. Причины
Синдром Миллера-Дикера был открыт в 1983 году. Основной причиной развития этого синдрома является делеция части хромосомы 17p13.3. Почти 100% пациентов с диагнозом синдрома Миллера-Дикера имеют эту делецию. Размер делеции варьируется среди индивидуумов. Около 50% имеют микроскопически видимые делеции, а остальные имеют субмикроскопические делеции. Клинические особенности синдрома Миллера-Дикера связаны с потерей нескольких генов в хромосоме 17. Потеря гена PAFAH1B1, как уже известно исследователям, отвечает за развитие лиссэнцефалии. Потеря гена YWHAE увеличивает тяжесть лиссэнцефалии, другие гены, потеря которых приводит к появлению других клинических особенностей этого синдрома, еще не известны.
Синдром Миллера-Дикера обычно не наследуется от родителей. Делеция, которая является результатом семейной хромосомной транслокации, встречается лишь у около 12% больных. Чаще всего эта делеция происходит случайно в процессе формирования половых клеток или на ранней стадии беременности. Вероятность иметь другого ребенка с этим же синдромом очень низкая (если эта делеция не будет связана с транслокацией).
Синдром Миллера-Дикера. Симптомы и проявления
Лиссэнцефалия, при синдроме Миллера-Дикера, характеризуется агирией / агирией с фронтальной пахигирией и толстой корой больших полушарий головного мозга (~ на 3 мм толще нормальной). Дополнительные особенности включают в себя заднее расширение боковых желудочков и гипоплазию (недоразвитие) мозолистого тела.

Ребенок с синдромом Миллера-Дикера
Синдром Миллера-Дикера связан с умственной отсталостью, задержками физического развития, характерными лицевыми особенностями, необычной мышечной спастичностью, трудностями в кормлении, низким мышечным тонусом (гипотония) и судорогами. Задержки развития, как правило, тяжелые и большинство детей не смогут научиться сидеть или ходить самостоятельно. Их развитие будет постоянно оставаться на уровне развития здорового 3-6 месячного ребенка. Трудности с кормлением и глотанием являются общими и они могут осложняться аспирационной пневмонией. Низкий мышечный тонус (гипотония) является характерной особенностью этой аномалии и, как правило, она отмечается по всему телу. Приступы обычно начинаются в период между рождением и 6 месяцами. Окружность головы маленькая. Чем сильнее будут сглажены извилины, тем тяжелее будут симптомы и проявления. Характерные лицевые особенности включают: выпуклый лоб, затонувшее лицо, небольшой вздернутый нос, аномальные и низко расположенные уши, небольшие челюсти, толстая верхняя губа. Некоторые дети с этим синдромом растут медленнее, чем их здоровые сверстники. У некоторых детей отмечаются пороки развития сердца / почек и пупочная грыжа.
Синдром Миллера-Дикера. Диагностика
Беременность, осложненная плодом с синдромом Миллера-Дикера, может быть связана с многоводием (избыток амниотической жидкости), задержкой внутриутробного роста и снижением движений плода. Постановка диагноза и его подтверждение основывается на хромосомном анализе. Классическая лиссэнцефалия, как правило, становится видимой (при проведении МРТ) только с 28 недели беременности.
Синдром Миллера-Дикера. Лечение
Лечение только симптоматическое и зависит от тяжести симптомов. Контроль приступов имеет очень важное значение. Питательная трубка может быть использована для решения проблем с кормлением. Трудотерапия, физиотерапия и специальные образовательные программы могут помочь ребенку достичь потенциала на столько, на сколько это возможно.
Синдром Миллера-Дикера. Прогноз
Прогноз развития варьируется в зависимости от тяжести пороков развития головного мозга. Большинство детей умирают в течение первых нескольких лет жизни. Тем не менее, с улучшенным контролем приступов и с использованием питательной трубки, дети с этим синдромом живут дольше. Меньшинство пациентов с синдромом Миллера-Дикера доживают до раннего подросткового возраста. Аспирационная пневмония является наиболее распространенной причиной наступления смерти.
Источник
Синдром Миллер-Дикер (сокращенно МДС ), Миллер-Дикер Лиссэнцефалия синдром (MDLS) и синдром хромосомы 17p13.3 удаления является синдром микро удаления характеризуется врожденными пороками развития . Врожденные пороки развитие являются физическими дефектами выявляемых у ребенка при рождении , который может включать в себя множество различных частей тела , включая мозг, сердце, легкие, печень, кости, или желудочно — кишечный тракт. МДС представляет собой синдром смежных генов — расстройство в связи с удалением нескольких локусов генов , прилегающих друг к другу. Заболевание возникает в результате делеции части небольшого плеча хромосомы 17p (который включает в себя как Lis1 и 14-3-3 эпсилоне генов), что приводит к частичной моносомии . Там может быть несбалансированные транслокации (т.е. 17Q: 17p или 12q: 17p), или наличие кольцевой хромосомы 17.
Этот синдром не следует путать с синдромом Миллера , несвязанной редким генетическим расстройством, или синдром Миллера Фишера, форма синдрома Гийена-Барре .
Характеристики
Мозг аномально гладкий, с меньшим количеством складок и пазами. Лицо, особенно у детей, имеет различные характеристики, включая короткий нос с загнутыми ноздрями, утолщенного верхней губой с тонкой алой верхней границей, лобной начальствование, небольшой челюстью, низкими posteriorily повернутых ушами, затонувшим появлением в середине лица, широко расставленные глаза, и гипертелоризм. Лоб видным с битемпоральными выдалбливать.
Характеристики, которые не включают в себя визуальный умственная отсталость, до и послеродовое замедление роста, эпилепсия, и снижение продолжительности жизни.
Неспособность процветать , кормя трудности, судороги и снижение спонтанной активности часто наблюдаются. Смерть обычно происходит в младенчестве и детстве. Могут возникать множественные аномалии головного мозга, почек и желудочно — кишечного тракта (желудка и кишечника).
Причина и генетика
МДС представляет собой синдром микроделеционного , включающее потерю гена pafah1b1 на хромосоме 17 , который отвечает за характерный признак синдрома о Лиссэнцефалии. Потери другого гена, YWHAE , в одной и той же области хромосомы 17 увеличивает тяжесть Лиссэнцефалия у больных с синдромом Миллера-Дикера. Дополнительные гены в удаленной области могут внести свой вклад в различные особенности синдрома Миллер-Дикер.
Это может быть случайным событием в процессе формирования половых клеток или в раннем развитии плода или из — за семейной хромосомной перестройкой называемой хромосомной транслокации. В менее чем на 20%, наследование через аутосомно — доминантный тип. Родитель, как правило , не влияет, но несет определенную хромосомную перестройку , называемую сбалансированную транслокацию , в котором ни генетический материал не получил или потерял. Увеличение скорость необъяснимой потери плода может наблюдаться в МДСЕ носителях с сбалансированными транслокациями , хотя они могут быть иначе бессимптомно. Тем не менее, они могут стать также несбалансированным , поскольку они передаются следующему поколению. Синдром Миллера-Дикера, как правило , не наследуется. Событие удаления происходит случайно во время гаметогенеза (образование яиц или спермы) или в раннем развитии плода. Поэтому никакой истории заболевания не обычно видели в своих семьях.
диагностика
Болезнь может быть диагностирована цитогенетическими методами , как флуоресценции в гибридизация (FISH), тестирование на микроделеция в LIS1.
Раннее обнаружение
При использовании пренатального ультразвукового изображения, раннее обнаружение аномального развития мозга у плода с МДСОМ можно увидеть. При рождении дисморфизм лиц может присутствовать в младенце. Маленькие дети, когда страдают, могут страдать от кормления трудностей, тяжелой умственной отсталости, задержки развития и эпилептических припадков. МРТ способствует раннему выявлению этого синдрома у детей, выявляя «гладкий мозг» образ, называемый также Лиссэнцефалию . Дети с этим синдромом могут оставаться гиподиагностика из — за редкости и распространенности черты лица , которые кажутся дисморфозом. Синдром разделяет отдельные внешние особенности ( фенотип ) , сходные с более общими синдромами. Отсутствие соответствующей семейной истории может задержать диагноз. FDNA предоставляет услугу , которая в свою очередь , увеличивает вероятность обнаружения этих различных характеристик, которые, когда сведен к генетику, может помочь в достижении правильного медицинского диагноза. Если пара имеет один ребенок с МДС, они могут быть предложены пренатального скрининга в будущих беременностей. Этот параметр особенно важен для 20% МДС семей , где один из родителей несут сбалансированную хромосомную перестройку. Риск этих пар , имеющих одного ребенка с МДС , зависит от конкретного типа хромосомной перегруппировки настоящего и может достигать 25-33%. Для семей , в которых хромосомы обоих родителей являются нормальными, риск наличия другого ребенка с МДС является низким (1% или меньше). Либо хорион (CVS) или амниоцентез может быть использован на ранней стадии беременности , чтобы получить небольшой образец клеток от развивающегося эмбриона для исследования хромосом. Ранняя пренатальная диагностика с помощью ультразвука не является надежной , потому что мозг , как правило , гладко до поздних сроков беременности. Пары , которые рассматривают диагноз пренатальной должны обсудить риски и преимущества данного типа тестирования с генетиком или генетическим консультантом.
Визуальный мозг
Мозг, как правило, грубо ненормальным в общих чертах, когда кто-то с диагнозом синдром Миллера-Дикера. Только несколько мелких борозд и мелкие латеральной трещины видно; это берет на песочные часы или фигура-8 появления на осевой томографии. Толщина и измерение для человека без МДС составляет 3-4 мм. С МДС, кора головного мозга человека измеряется на 12-20 мм.
лечение
Хотя никакого лечения МДС не доступна, многие осложнения, связанные с этим условием можно лечить, и многое может быть сделано, чтобы поддержать или компенсировать функциональные инвалидности. Из-за разнообразия симптомов, это может быть необходимо, чтобы увидеть целый ряд различных специалистов и пройти различные обследования, в том числе:
- Развивающая оценка
- оценка Кардиологи
- отоларингология
- Лечение судорог
- Урологическое оценка
- Генетическое консультирование сбалансированного хромосомная транслокация должна быть исключена в течение родителей с больным ребенком планирует еще одну беременности, поэтому родители с больными детьми следует посетить генетическую консультацию.
Прогноз
Большинство людей с этим заболеванием не доживает детства. Лица с МДСОМ обычно умирают в младенчестве, и поэтому не доживают до возраста, где они могут размножаться и передавать МДС своего потомства.
эпидемиология
Миллер-Дикер происходит менее чем один в 100000 людей и может произойти во всех гонках.
история
МДС был назван в честь двух врачей, Джеймс Q. Миллера и Г. Дикера., Которые независимо друг от друга описали состояние в 1960 — х годах. Признаком МДС является Лиссэнцефалия, условие , в котором наружный слой мозга, кора головного мозга, аномально толстый и не хватает нормальных извилины (извилины). В некоторых областях мозга, извилины в меньшем количестве , но шире , чем обычно (pachygyri). Другие области не хватает извилин полностью (agyri). Как правило, в течение третьего и четвертого месяца беременности, клетки головного мозга в ребенке размножаться и двигаться к поверхности мозга, образуя кору. Лиссэнцефалия вызвана недостаточностью этого нервные клетки миграции. МДС часто называют Миллер-Дикеру Лиссэнцефалия синдром.
JQ Миллер описал болезнь, а в 1969 H Дикера подчеркнул, что он также должен взять синдром имени Лиссэнцефалии, поскольку некоторые пороки развития происходит за пределами самого мозга. Когда МДС была первоначально описана, генетики предположили, что это последовало аутосомно-рецессивный тип наследования. В начале 1990-х годов было обнаружено несколько пациентов с синдромом Миллера-Дикера, чтобы пропускать небольшую часть хромосомы 17. (17p13.3) (частичное удаление).
Рекомендации
внешняя ссылка
- Миллер-Дикер
- Синдром Миллера-Дикера
Источник