D68 код по мкб 10

Связанные заболевания и их лечение
Описания заболеваний
Национальные рекомендации по лечению
Стандарты мед. помощи
Содержание
- Синонимы диагноза
- Описание
- Симптомы
- Причины
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
D68,0 Болезнь Виллебранда.
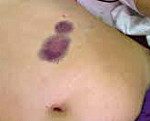
D68.0 Болезнь Виллебранда
Синонимы диагноза
Болезнь виллебранда, болезнь виллебранда-юргенса, ангиогемофилия, виллебранда-юргенса конституциональная тромбопатия, синдром виллебранда-юргенса.
Описание
Болезнь Виллебранда — наследственное заболевание крови, характеризующееся возникновением эпизодических спонтанных кровотечений, которые схожи с кровотечениями при гемофилии. Заболевание наследуется по принципу аутосомного доминирования.
Впервые данное заболевание было изучено в 1926 г. Ученым Виллебрандом, который на Аландских островах описал семью со своеобразным аутосомно-доминантно наследуемым геморрагическим диатезом, сходным как с тромбоцитовазопатией, так и с гемофилией. Было отмечено, что члены данной семьи страдают классической формой болезни, принадлежащей к I типу. Подтверждено доминантное наследование с разной проявляемостью патологического гена. Этим болезнь Виллебранда отличается от гемофилии, которая жестко наследуется и при которой у всех болеющих членов семьи в разных поколениях отмечается одна и та же выраженность дефицита фактора VIII.
По распространенности среди всех наследственных геморрагических диатезов болезнь Виллебранда занимает 3-е место после тромбоцитопатий и гемофилии А.
D68.0 Болезнь Виллебранда
Симптомы
Выраженность геморрагического синдрома при болезни Виллебранда варьирует от весьма легких форм с редко наблюдающимися носовыми кровотечениями и небольшими кровоизлияниями в кожу до крайне тяжелых вариантов с очень частыми, длительными и обильными кровотечениями самой разнообразной локализации, формированием гематом и больших кровоизлияний в мягких тканях и во внутренних органах. Иногда возникают кровоизлияния в суставы.
Геморрагический синдром при I типе намного тяжелее, чем при IIА и IIB типах болезни.
Следует отметить, что интенсивность кровотечений самой различной локализации (желудочно-кишечных, маточных, носовых) зачастую не соответствует нарушению коагуляциоиного и сосудисто-тромбоцитарного гемостазов.
В частности, на фоне умеренных нарушений в этих звеньях гемостаза упорно повторяются катастрофические кровотечения какой-либо одной локализации. В подобных случаях следует думать о каких-то дополнительных местных сосудистых или стромальных дисплазиях, провоцирующих кровотечения. Для их выявления необходимо тщательное дополнительное исследование слизистых оболочек носа, зева и глотки, ротовой полости, желудка и кишечника (риноскопия, ларингоскопия, фибродуоденогастроскопия, колоноскопия). На слизистых оболочках нередко обнаруживаются сосудистые образования в виде поверхностно расположенных расширенных и извитых сосудов диаметром 1-2 мм, легко дающих обильные, трудно останавливаемые кровотечения.
Известно, что такие сосудистые образования, чаще — артериовенозные соустья, служат причиной повторяющихся желудочно-кишечных кровотечений. Данные соустья наиболее опасны при болезни Виллебранда, когда нарушены основные механизмы купирования кровотечений.
Следует отметить, что сочетание болезни Виллебранда с ангиодисплазиями и другими дефектами соединительной ткани нельзя считать случайным. У больных с этим заболеванием часто выявляется пролабирование створок митрального и других клапанов сердца, ошибочно диагностируемое без эхокардиографии как ревматический митральный порок сердца.
В таком же свете следует рассматривать сочетания болезни Виллебранда с гиперэластозом кожи, слабостью связочного аппарата (частые и привычные вывихи, разболтанность суставов, реже — синдром Марфана).
Интенсивность кровотечения имеет прямую зависимость от уровня фактора VIII в плазме, что следует учитывать как при травмах, так и при хирургических вмешательствах.
При постановке диагноза заболевания Виллебранда основываются на совокупности следующих признаков: аутосомно-доминантном наследовании заболевания, кровоточивости, значительном удлинении времени кровотечения.
К основным диагностическим признакам добавляется ряд важных функциональных характеристик, облегчающих распознавание редуцированных вариантов болезни Виллебранда.
При болезни Виллебранда выявляется постепенное, а не немедленное нарастание активности фактора VIII в плазме больных после трансфузии антигемофильной плазмы.
Коррекционный эффект трансфузии намного превосходит количество вводимого фактора VIII.
Примечательна значительно большая, чем при гемофилии, продолжительность эффекта однократного переливания — около 36 ч, что характеризуется большей продолжительностью жизни в циркуляции реципиента фактора VIII.
Наиболее информативно количественное определение фактора Виллебранда в плазме больного.
Перечисленные диагностические признаки позволяют в большинстве случаев четко аргументировать диагноз болезни Виллебранда, определять ее тяжесть и форму.
Капилляроскопические изменения (неравномерность и закрученность петель капилляров, их булавовидные расширения) в диагностике болезни Виллебранда не используют, поскольку они выявляются менее чем у половины больных и далеко не патогномоничны, однако совместно с этим они весьма демонстративны и способствуют правильной диагностике.
Аутосомно-рецессивная форма болезни Виллебранда является самостоятельным заболеванием. У гетерозигот заболевание протекает совсем или почти бессимптомно, тогда как у гомозигот наблюдается крайне тяжелая кровоточивость при почти полном отсутствии фактора VIII в плазме. Однако поражение суставов и других частей опорно-двигательного аппарата, несмотря на такой значительный дефицит фактора VIII, все же гораздо легче, чем при гемофилии.
Причины
В основе патогенетических механизмов болезни Виллебранда лежит нарушение синтеза основного крупномолекулярного компонента фактора VIII, называемого также фактором Виллебранда.
По вышеперечисленным характеристикам различают «классический» тип болезни Виллебранда (тип I), при котором имеется более или менее выраженный парез синтеза рассматриваемого фактора с конкордантным снижением в плазме всех его компонентов и соответствующим отсутствием или уменьшением содержания в сосудистом эндотелии.
От этого типа отличаются вариантные формы болезни (тип II), при которых имеются качественные аномалии мультимерной структуры и свойств компонентов фактора VIII.
Болезнь по доминированию дефицита компонентов фактора VIII причисляется к плазменным дефектам гемостаза. При более углубленном рассмотрении с неменьшим правом ее можно отнести к первично-сосудистым заболеваниям, поскольку в основе болезни лежит ослабление или извращение синтеза фактора Виллебранда в эндотелии кровеносных сосудов — единственном месте его образования в организме.
Старое название болезни «ангиогемофилия» весьма близко к правильному пониманию ее сути, хотя в настоящее время редко употребляется.
Лечение
Наиболее важным патогенетическим моментом в терапии, способствующим нормализации (зачастую только временной) всех нарушенных гемостатических функций, является трансфузионная терапия – введение гемопрепаратов, содержащих комплекс фактора VIII, в том числе фактор Виллебранда.
С этой целью чаще всего используют антигемофильную плазму и криопреципитат.
Кроме того, под влиянием введенного извне фактора Виллебранда повышается собственная продукция фактора VIII, поэтому заместительная терапия болезни Виллебранда требует значительно более редких трансфузий и меньших доз гемопрепаратов, чем лечение гемофилии А. Однократные переливания антигемофильной плазмы или криопреципитата повышают к концу первых суток уровень фактора VIII почти до 100%, после чего его концентрация выше 50% поддерживается самостоятельно в течение 36 Правда, концентрация самого фактора Виллебранда снижается раньше, в силу чего тромбоцитарно-сосудистый гемостаз вновь нарушается при еще высоком уровне VIIIk в плазме. Этим объясняется то, что при болезни Виллебранда трансфузионная терапия более стойко и надежно поддерживает уровень фактора VIII и предупреждает послеоперационные кровотечения, чем купирует микроциркуляторные кровотечения (маточные и носовые). В связи с этим наиболее целесообразно введение гемопрепаратов (антигемофильной плазмы, криопреципитата) не реже 1 раза в 2 дня и в разовой дозе не меньше 15 ЕД/кг.
Коррекция развивается постепенно, поэтому перед хирургическими вмешательствами трансфузии начинают за 2-4 дня до операции, а при родах – в самом начале родовой деятельности.
Показанием к заместительной терапии служат обильные и длительные кровотечения любой локализации, хотя при маточных кровотечениях такое лечение не всегда эффективно.
Переливания тромбоцитной массы при болезни Виллебранда неэффективны, поскольку дисфункция тромбоцитов при этой болезни вторична. По этой же причине при болезни Виллебранда оказались малоэффективными и многие фармакологические препараты, применяющиеся при тромбоцитопатиях, — АТФ, соли магния, серотонин и.
Принципиально новым является использование в лечении болезни Виллебранда аргинин-терминального синтетического аналога вазопрессина.
При легких и среднетяжелых заболеваниях доказана эффективность аминокапроновой кислоты в дозах до 0,2 г/(кг/сут), которая применяется при всех кровотечениях микроциркуляторного типа, в том числе при маточных кровотечениях с первого дня менструального цикла до окончания менструации.
Следует избегать совместного применения аминокапроновой кислоты, противозачаточных препаратов и криопреципитата, так как такое лечение может осложниться диссеминированным внутрисосудистым свертыванием крови или тромбозами. Носовые кровотечения купируют так же, как при гемофилии.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
Рубрика МКБ-10: D68.8
МКБ-10 / D50-D89 КЛАСС III Болезни крови, кроветворных органов и отдельные нарушения, вовлекающие иммунный механизм / D65-D69 Нарушения свертываемости крови, пурпура и другие геморрагические состояния / D68 Другие нарушения свертываемости
Определение и общие сведения[править]
Сочетанный дефицит факторов V и VIII
Комбинированный дефицит факторов V и VIII — редкое аутосомно-рецессивное нарушение. Обычно встречается как следствие близкородственных браков.
Распространенность оценивается в интервале от 1/100000 и 1/1000000.
Этиология и патогенез[править]
Это нарушение вызывается мутациями либо гена LMAN1 (хромосома 18; Q21) или гена MCFD2 (хромосома 2).
LMAN1 кодирует белок ERGIC-53, который является трансмембранным лектином, в то время как MCFD2 является EF-рукасодержащим протеином. Белковый комплекс ERGIC-53/MCFD2 функционирует как грузовой рецептор, который облегчает транспортировку факторов свертывания V и VIII из эндоплазматического ретикулума к аппарату Гольджи.
Мутации в LMAN1 происходят в 70% случаев и включают в себя только нулевые мутации. Мутации гена MCFD2 происходят в около 30% случаев и включают в себя как нулевые и миссенс мутации.
Клинические проявления[править]
У пациентов с данной формой коагулопатии отмечают редкие эпизоды носовых кровотечений и незначительную склонность к развитию поверхностных синяков. Уровень факторов V и VIII у них обычно в пределах, достаточных для гемостаза. Поэтому тяжелых спонтанных кровотечений, как правило, не бывает. Однако для этих людей характерны кровотечения после оперативных вмешательств, в том числе после экстракции зуба, в связи с травмой. У женщин описаны меноррагии и кровотечения в послеродовом периоде. Эти кровотечения по тяжести обычно соответствуют таковым у больных с изолированным дефицитом фактора V или VIII.
Другие уточненные нарушения свертываемости: Диагностика[править]
Комбинированный дефицит факторов V и VIII связан с удлинением АПТВ и протромбинового времени. При этом степень удлинения АПТВ обычно превосходит изменение последнего показателя. Это можно объяснить тем, что оба фактора принимают участие в реакции активации свертывания по внутреннему пути, для оценки которой используется АПТВ. Изменение протромбинового времени в данном случае будет зависеть исключительно от уровня фактора V.
Дифференциальный диагноз[править]
Дифференциальная диагностика включает в себя мягкую фору гемофилии А и частичный дефицит фактора V.
Другие уточненные нарушения свертываемости: Лечение[править]
Лечение геморрагических эпизодов зависит от локализации кровотечения и уровня дефицитных факторов. Обычно используют концентраты фактора VIII, а также СЗП (источник фактора V). Интервалы между трансфузиями составляют 12 ч. При умеренных кровотечениях уровень VIII должен быть повышен до 30%, а при тяжелых кровотечениях и во время оперативных вмешательств — до 50%. Уровень фактора V при введении СЗП повышают до 25%, что обычно достаточно для гемостаза.
Прогноз
Прогноз благоприятный для умеренных форм заболевания. Лечение больных с более тяжелыми формами следует проводить в специализированном центре.
Профилактика[править]
Прочее[править]
Врожденный дефицит ингибитора активатора плазминогена 1-го типа
Определение и общие сведения
Врожденный дефицит ингибитора активатора плазминогена 1-го типа является редким генетическим заболеванием, которое характеризуется преждевременныс лизисом гемостатических сгустков гемостаза и умеренной тенденцией к кровотечениям.
Распространенность и заболеваемость неизвестны. Частичный и полной дефицит ингибитора активатора плазминогена являются чрезвычайно редкими заболеваниями. В общине амишей обнаружено восемнадцать гомозиготных пациентов с клиническими симптомами заболевания и более чем 100 гетерозиготных пациентов без признаков кровотечений было зарегистрировано на сегодняшний день.
Этиология и патогенез
Ингибитор активатора плазминогена 1-го типа является физиологическим ингибитором тканевого активатора плазминогена, основного источника внутрисосудистого фибринолиза. Дефицит может быть качественным или количественным, у нескольких пациентов белок присутствует, но является функционально неактивными. Пострадавшие пациенты несут один (гетерозиготы) или два (гомозиготы) аллеля с мутацией гена SERPINE1 (7q22.1), что приводит к частичному или полному антигенному дефициту ингибитора активатора плазминогена 1-го типа.
Частичный и полный дефициы передаются как аутосомно-рецессивные признаки.
Клинические проявления
Клинические признаки дефицита могут проявиться в раннем младенческом возрасте. Спонтанные кровотечение наблюдаются редко. Часто наблюдаются кровоподтеки или умеренные геморрагии, локализующиеся в суставах (колени, локти), носу и деснах, которые, как правило, вызываются легкими травмами. Менструальные кровотечения могут быть обильными и нередки длительное кровотечения после хирургических вмешательств. Кровотечения менее частые и менее серьезные (или их отсутствие) отмечаются у гетерозиготных особей (частичное покрытие дефицита) и эти симптомы могут появиться в конце жизни после травмы или хирургического вмешательства.
Диагностика
Диагноз основывается на проведении антигенных и функциональных (тест ингибирования активатора плазминогена ) анализов. Анализ генотипа может быть необходимым для проведения семейного исследования. Молекулярно-генетическое исследование подтверждает диагноз.
Дифференциальный диагноз
Дифференциальная диагностика включает в себя приобренный дефицит ингибитора активатора плазминогена 1-го типа и дефицит альфа2-антиплазмина.
Лечение
Эффективны ингибиторы фибринолиза (эпсилонаминокапроновая кислота или транексамовая кислота), необходимо избегать использования переливаний крови и ее производных. Менструации и беременность требует особого внимания в отношении диагностики и лечения.
Прогноз
Прогноз, как правило, хороший. Кровотечение можно предотвращать и контролировать с помощью антифибринолитической терапии.
Источники (ссылки)[править]
Гематология [Электронный ресурс] : национальное руководство / под ред. О.А. Рукавицына — М. : ГЭОТАР-Медиа, 2015. — https://www.rosmedlib.ru/book/ISBN9785970433270.html
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
- Аминокапроновая кислота
Источник
Исключены:
- отдельные состояния, возникающие в перинатальном периоде (P00-P96)
- болезни височно-нижнечелюстного сустава (K07.6)
- некоторые инфекционные и паразитарные болезни (A00-B99)
- синдром сдавления (T79.6)
- осложнения беременности, родов и послеродового периода (O00-O99)
- врожденные аномалии, деформации и хромосомные нарушения (Q00-Q99)
- болезни эндокринной системы, расстройства питания и нарушения обмена веществ (E00-E90)
- травмы, отравления и некоторые другие последствия воздействия внешних причин (S00-T98)
- новообразования (C00-D48)
- симптомы, признаки и отклонения от нормы, выявленные при клинических и лабораторных исследованиях, не классифицированные в других рубриках (R00-R99)
Этот класс содержит следующие блоки:
- M00-M25 Артропатии
- M00-M03 Инфекционные артропатии
- M05-M14 Воспалительные полиартропатии
- M15-M19 Артрозы
- M20-M25 Другие поражения суставов
- M30-M36 Системные поражения соединительной ткани
- M40-M54 Дорсопатии
- M40-M43 Деформирующие дорсопатии
- M50-M54 Другие дорсопатии
- M60-M79 Болезни мягких тканей
- M60-M63 Поражения мышц
- M65-M68 Поражения синовиальных оболочек и сухожилий
- M70-M79 Другие поражения мягких тканей
- M80-M94 Остеопатии и хондропатии
- M80-M85 Нарушения плотности и структуры кости
- M86-M90 Другие остеопатии
- M91-M94 Хондропатии
- M95-M99 Другие нарушения костно-мышечной системы и соединительной ткани
Звездочкой отмечены следующие категории:
- M01* Прямое инфицирование сустава при инфекционных и паразитарных болезнях, классифицированных в других рубриках
- M03* Постинфекционные и реактивные артропатии при болезнях, классифицированных в других рубриках
- M07* Псориатические и энтеропатические артропатии
- M09* Ювенильный артрит при болезнях, классифицированных в других рубриках
- M14* Артропатии при других болезнях, классифицированных в других рубриках
- M36* Системные поражения соединительной ткани при болезнях, классифицированных в других рубриках
- M49* Спондилопатии ткани при болезнях, классифицированных в других рубриках
- M63* Поражения мышц при болезнях, классифицированных в других рубриках
- M68* Поражения синовиальных оболочек и сухожилий при болезнях, классифицированных в других рубриках
- M73* Поражения мягких тканей при болезнях, классифицированных в других рубриках
- M82* Остеопороз при болезнях, классифицированных в других рубриках
- M90* Остеопатии при болезнях, классифицированных в других рубриках
ЛОКАЛИЗАЦИЯ КОСТНО-МЫШЕЧНОГО ПОРАЖЕНИЯ
В классе XIII для обозначения локализации поражения введены дополнительные знаки, которые могут факультативно использоваться с соответствующими подрубриками. Поскольку место распространения или специальная адаптация могут варьироваться в количестве используемых цифровых характеристик, предполагается, что дополнительная подклассификация по локализации должна быть помещена в идентифицируемую отдельную позицию (например, в дополнительный блок). Различные подклассификации, используемые при уточнении повреждения колена, дорсопатиях или биомеханических нарушениях, не классифицированных в других рубриках, приведены в M23, в M40-M43 и в M99 соответственно
- 0 Множественная локализация
- 1 Плечевая область
- Ключица,
- акромиально-ключичный сустав,
- лопатка,
- плечевой сустав,
- грудино-ключичный сустав
- 2 Плечо
- Плечевая кость
- Локтевой сустав
- 3 Предплечье
- Лучевая кость
- Лучезапястный сустав,
- локтевая кость
- 4 Кисть
- Запястье,
- Суставы между этими костями
- пальцы,
- пясть
- 5 Тазовая область и бедро
- Ягодичная область
- Тазобедренный сустав,
- крестцо-подвздошный сустав
- бедренная кость,
- таз
- 6 Голень
- Малоберцовая кость,
- большеберцовая кость
- Коленный сустав
- 7 Голеностопный сустав и стопа
- Голеностопный сустав,
- Плюсна,
- предплюсна,
- другие суставы стопы пальцы стопы
- 8 Другие
- Голова, шея, ребра, череп, туловище, позвоночник
- 9 Локализация неуточненная
последние изменения: январь 2004
Следующие дополнительные пятые знаки, обозначающие локализацию поражения, даны для факультативного использования с соответствующими рубриками блока «Дорсопатии», исключая рубрики M50 и M51; см. также примечание в разделе M00-M99.
- 0 Множественные отделы позвоночника
- 1 Область затылка, первого и второго шейных позвонков
- 2 Область шеи
- 3 Шейно-грудной отдел
- 4 Грудной отдел
- 5 Пояснично-грудной отдел
- 6 Поясничный отдел
- 7 Пояснично-крестцовый отдел
- 8 Крестцовый и крестцово-копчиковый отдел
- 9 Неуточненная локализация
Источник