Что такое синдром smith magenis

Синдром Смит-Магенис (СМС) — наследственное заболевание, имеет такие проявления, как умственная отсталость, аномалии лица, проблемы со сном и многочисленные поведенческие проблемы, в том числе и такие, как самоповреждение. Частота синдрома Смит-Магенис составляет 1 случай на 15 000-25 000 человек[1]. Синдром Смит-Магенис, как правило, обусловлен микроделецией в коротком плече 17-ой хромосомы (17p), и иногда его называют 17p-синдром[2].
Симптомы[править | править код]
У маленького ребёнка с синдромом Смит-Магенис череп и мягкие ткани головы имеют следующие особенности: широкое и квадратное лицо, глубоко посаженные глаза, большие щеки и выступающую челюсть, а также плоская переносица. По мере взросления ребёнка переносица становится более выраженной, «трамплиновидной». Глаза, как правило, глубоко и близко друг к другу посажены. Брови широкие, густые, сросшиеся, расширяющиеся по бокам. Рот — самая заметная особенность, верхняя и нижняя губы полные, рот широкий. Рот изогнут вниз, а верхняя губа приподнята наружу, благодаря мясистому губному желобку. Эти черты лица становятся более заметными с возрастом, так как рост нижней челюсти опережает рост верхней, что приводит к явной гипоплазии средней части лица. Присутствует также лёгкая брахицефалия[3].
Для пациентов с синдромом Смит-Магенис с раннего возраста характерны нарушения сна. Больные могут быть очень сонными в течение дня, но испытывают проблемы с засыпанием и просыпаются несколько раз каждую ночь из-за инвертированного циркадного ритма выработки мелатонина[4].
Люди с синдромом Смит-Магенис очень общительны и, как правило, ласковы, но все они имеют множество поведенческих проблем. Поведенческие проблемы заключаются в частых вспышках гнева, агрессии, беспокойстве, компульсивном поведении, импульсивности и трудностях с обращением внимания. Самоповреждения, в том числе укусы, удары, удары по голове и ковыряние, очень распространены. Повторяющиеся самостоятельные обнимания — это поведенческая черта, которая может быть уникальной для синдрома Смит-Магенис. Люди с этим заболеванием могут также принудительно облизывать пальцы и перелистывать страницы книг и журналов (поведение, известное как «лизать и переворачивать»), а также обладать впечатляющей способностью вспомнить широкий спектр мелких деталей о людях или предметной области, пустяки.
Другие симптомы могут включать низкий рост, искривление позвоночника (сколиоз), хриплый голос, снижение чувствительности к боли и температуре. У некоторых людей с этим расстройством есть нарушения слуха, которые приводят к глухоте. Пострадавшие люди могут иметь нарушения зрения, которые вызывают близорукость, косоглазие и другие проблемы со зрением. У людей с синдромом Смит-Магенис также отмечались пороки сердца и почек, хотя они встречаются редко.
Генетика[править | править код]
Синдром Смит-Магенис в большинстве случаев вызван хромосомной перестройкой, приводящей к утрате определённых сегментов одной хромосомой из пары 17 хромосом[5]. У большинства людей с СМС происходит делеция генетического материала из определённой области 17 хромосомы (17p11.2). Хотя этот регион содержит несколько генов, недавно исследователи обнаружили, что потеря конкретного гена RAI1 отвечает за большинство характерных особенностей этого синдрома[6][7]. Утрата соседних с RAI1 генов в хромосоме 17 усугубляет серьёзность клинических признаков и приводит к вариабельности проявлений синдрома. Потеря дополнительных генов в делетированном участке может помочь объяснить, почему особенности синдрома Смит-Магенис варьируют среди пораженных людей. У небольшой доли пациентов синдромом Смит-Магенис вызван мутацией в гене RAI1, а не делецией хромосомы 17.
Делеции хромосомы 17 и мутации гена RAI1 приводят к выработке аномальной или нефункциональной версии белка RAI1. RAI1 является транскрипционным фактором, который регулирует экспрессию множества генов, в том числе нескольких, которые участвуют в контроле циркадного ритма, таких как CLOCK[8]. Группы, возглавляемые Джеймсом Лупски (Медицинский колледж Бэйлора) и Сарой Элси (Университет Содружества Вирджинии), в настоящее время изучают роль этого гена в патогенезе синдрома Смит-Магенис[9][10].
СМС обычно не наследуются. Это состояние обычно возникает в результате мутации, происходящей во время образования репродуктивных клеток (яйцеклеток или сперматозоидов) или в начале эмбрионального развития. Люди с синдромом Смит-Магенис чаще всего не имеют истории заболевания в своей семье.
Диагностика[править | править код]
Синдром Смит-Магенис обычно подтверждается анализом клеток крови, который называют цитогенетическим анализом, или кариотипированием. Для диагностики используется метод, называемый FISH (флуоресцентная гибридизация in situ). Делеции небольшого размера (микроделеции) иногда оставались незамеченными в стандартном тесте FISH, что приводило к отрицательныму результату кариотипирования у пациентов с СМС.
Недавняя разработка теста FISH на делецию в 17p11.2 позволила более точно обнаруживать это нарушение[11]. Однако требуется дальнейшее разработка диагностики для выявления синдрома Смит-Магенис, вызванного мутацией гена RAI1.
Детям с СМС часто ставят психиатрические диагнозы, такие как аутизм, синдром дефицита внимания и гиперактивности (СДВГ), обсессивно-компульсивное расстройство (ОКР), синдром дефицита внимания (СДВГ) и / или расстройства настроения.
Лечение[править | править код]
Лечение синдрома Смит-Магенис основано на управлении его симптомами. Дети с СМС часто нуждаются в нескольких формах поддержки, включая физиотерапию, трудотерапию и логопедию. Поддержка часто требуется в течение всей жизни больного.
Лекарства часто используются для устранения некоторых симптомов. Мелатониновые добавки и тразодон обычно используются для регуляции нарушений сна. В сочетании с экзогенным мелатонином блокада выработки эндогенного мелатонина в течение светлого времени суток адренергическим антагонистом ацебутололом может повысить концентрацию, улучшить время сна и время сна и помочь в улучшении поведения[12]. Другие лекарства (такие как рисперидон) иногда используются для регулирования агрессивного поведения.
Происхождение названия[править | править код]
Название «Смит-Магенис» происходит от фамилий двух женщин-учёных, которые в 1986 году описали это генетическое заболевание. Это Энн С. М. Смит, генетический консультант в Национальных институтах здравоохранения, и Рут Эллен Магенис, педиатр, медицинский генетик и цитогенетик из Орегонского университета наук о здоровье[13][14].
Примечания[править | править код]
- ↑ Smith-Magenis syndrome (декабрь 2013). Дата обращения 27 марта 2016.
- ↑ Bi, W; Yan, J; Stankiewicz, P; Park, SS; Walz, K; Boerkoel, CF; Potocki, L; Shaffer, LG; Devriendt, K; Nowaczyk, MJ; Inoue, K; Lupski, J. R. Genes in a refined Smith-Magenis syndrome critical deletion interval on chromosome 17p11.2 and the syntenic region of the mouse. (англ.) // Genome Research (англ.)русск. : journal. — 2002. — May (vol. 12, no. 5). — P. 713—728. — doi:10.1101/gr.73702. — PMID 11997338.
- ↑ Allanson, Judith E.; Greenberg, Frank; Smith, Ann C. M. The face of Smith-Magenis syndrome: a subjective and objective study (англ.) // Journal of Medical Genetics (англ.)русск. : journal. — 1999. — Vol. 36. — P. 394—397.
- ↑ De Leersnyder H., De Blois M. C., Claustrat B., etal. Inversion of the circadian rhythm of melatonin in the Smith-Magenis syndrome (англ.) // J Pediatr (англ.)русск. : journal. — 2001. — Vol. 139, no. 1. — P. 111—116. — doi:10.1067/mpd.2001.115018. — PMID 11445803.
- ↑ Shaw, CJ; Withers, MA; Lupski, J. R. Uncommon deletions of the Smith-Magenis syndrome region can be recurrent when alternate low-copy repeats act as homologous recombination substrates. (англ.) // American Journal of Human Genetics (англ.)русск. : journal. — 2004. — July (vol. 75, no. 1). — P. 75—81. — doi:10.1086/422016. — PMID 15148657.
- ↑ Girirajan S., Vlangos C. N., Szomju B. B., etal. Genotype-phenotype correlation in Smith–Magenis syndrome: evidence that multiple genes in 17p11.2 contribute to the clinical spectrum (англ.) // Genet. Med. (англ.)русск. : journal. — 2006. — Vol. 8, no. 7. — P. 417—427. — doi:10.1097/01.gim.0000228215.32110.89. — PMID 16845274.
- ↑ Elsea, SH; Girirajan, S. Smith-Magenis syndrome. (англ.) // European Journal of Human Genetics (англ.)русск. : journal. — 2008. — April (vol. 16, no. 4). — P. 412—421. — doi:10.1038/sj.ejhg.5202009. — PMID 18231123.
- ↑ Williams S. R., Zies D., Mullegama S. V., etal. Smith-Magenis syndrome results in disruption of CLOCK gene transcription and reveals an integral role for RAI1 in the maintenance of circadian rhythmicity (англ.) // Am. J. Hum. Genet. (англ.)русск. : journal. — 2012. — Vol. 90, no. 6. — P. 941—949. — doi:10.1016/j.ajhg.2012.04.013. — PMID 22578325.
- ↑ Lupski, James Richard. Exploring the Reversibilty of the Smith-Magenis Syndrome Phenotype
- ↑ Building bridges of hope: taking stock of the Smith-Magenis Syndrome
- ↑ Lupski, James R.; Potocki, Lorraine; Chen, Ken-Shiung; Park, Sung-Sup; Osterholm, Doreen E.; Withers, Marjorie A.; Kimonis, Virginia; Summers, Anne M.; Meschino, Wendy S.; Anyane-Yeboa, Kwame; Kashork, Catherine D.; Shaffer, Lisa G. Molecular mechanism for duplication 17p11.2— the homologous recombination reciprocal of the Smith-Magenis microdeletion (англ.) // Nature Genetics : journal. — 2000. — 1 January (vol. 24, no. 1). — P. 84—87. — doi:10.1038/71743. — PMID 10615134.
- ↑ De Leersnyder, H. Inverted rhythm of melatonin secretion in Smith–Magenis syndrome: from symptoms to treatment (англ.) // Trends Endocrinol. Metab. (англ.)русск. : journal. — 2006. — September (vol. 17, no. 7). — P. 291—298. — doi:10.1016/j.tem.2006.07.007. — PMID 16890450.
De-Leersnyder H., de-Blois M. C., Vekemans M., Sidi D., Villain E., Kindermans C., Munnich A. β1-adrenergic antagonists improve sleep and behavioural disturbances in a circadian disorder, Smith–Magenis syndrome (англ.) // Journal of Medical Genetics (англ.)русск. : journal. — 2001. — September (vol. 38, no. 9). — P. 586—590. — doi:10.1136/jmg.38.9.586. — PMID 11546826. - ↑ synd/3884 на Who Named It?
- ↑ Smith A. C., McGavran L., Robinson J., etal. Interstitial deletion of (17)(p11.2p11.2) in nine patients (англ.) // Am. J. Med. Genet. (англ.)русск. : journal. — 1986. — Vol. 24, no. 3. — P. 393—414. — doi:10.1002/ajmg.1320240303. — PMID 2425619.
Ссылки[править | править код]
- Синдром Смит-Магенис. Сообщество родителей
- Синдром Смита-Магениса — этиология, клиника, диагностика на MedUniver
- Смит-Магенис cиндром, Центр молекулярной генетики
Источник
Кратко о синдроме
Синдром Смит—Магенис (Smith—Magenis syndrome) — это генетическое нарушение, возникающее при отсутствии небольшого участка 17-й хромосомы [обозначается как del(17р11.2)] и проявляющееся характерным строением тела, особенностями развития и поведения. Первая группа детей была описана в 1980 году в США врачом-клиницистом Энн Смит и цитогенетиком Эллен Магенис.
Симптомы:
— характерные черты лица (широкое квадратное лицо; короткоголовость, то есть относительно большие поперечные размеры головы; выпуклый лоб; сросшиеся брови; монголоидный разрез глаз; глубоко посаженные глаза; широкая переносица; недоразвитие средней трети лица; короткий нос с открытыми вперед ноздрями; малая высота носа; малые размеры нижней челюсти, а со временем — выступание нижней челюсти вперед; вывернутая наружу мясистая верхняя губа, имеющая форму шатра);
— низкий рост, маленькие кисти и стопы, короткие пальцы рук и ног, плоскостопие, необычная походка (с широко расставленными ногами);
— задержка физического развития;
— низкий мышечный тонус;
— проблемы вскармливания (выраженная орально-моторная дисфункция с нарушениями сосания и глотания, со слабостью мышц языка, с вытеканием слюны изо рта; отказ от пищи определенной консистенции, желудочно-пищеводный рефлюкс);
— низкий хриплый голос;
— хронический средний отит, тугоухость;
— затяжные риниты и синуситы, требующие лечения антибиотиками;
— косоглазие, близорукость и другая патология глаз;
— ослабленные рефлексы, признаки периферической нейропатии, пониженная чувствительность к боли;
— частые запоры;
— сколиоз;
— повышенный уровень холестерина в крови;
— задержка психического развития;
— умственная отсталость (легкая или средней тяжести);
— задержка речевого развития, проблемы с артикуляцией;
— дефицит внимания, часто в сочетании с гиперактивностью;
— нарушения сна (ночной сон менее глубокий, с частыми пробуждениями; ранние утренние подъемы; дневная сонливость), обусловленные неправильным суточным ритмом секреции мелатонина;
— стереотипное поведение: порывистое сжатие руками верхней половины тела (самообъятие); перелистывание страниц, слюнявя палец; стремление все засовывать в рот, в том числе собственные руки; скрежетание зубами;раскачивание туловища; верчение и кручение предметов;
— дезадаптивное поведение: частые вспышки гнева, требование внимания к себе со стороны взрослых, импульсивность, отвлекаемость, непослушание, агрессия, проблемы с отправлением естественных потребностей;
— склонность к самоповреждению (больные могут сами себя бить, кусать, щипать, вводить инородные тела в естественные отверстия тела, сдирать ногти на руках и ногах);
— реже встречаются патология сердца, почек и мочевых путей, нарушения функции щитовидной железы, эпилепсия, нарушения иммунитета, расщелина неба и расщелина губы, отслойка сетчатки.
При этом дети обычно ласковы, добродушны, общительны, любят внимание и поощрение, эмоциональны, легко подражают другим, любят музыку, обладают чувством юмора и хорошей долговременной памятью на имена, места и события. Диагноз нередко ставится только в школьном возрасте, когда становятся очевидными характерные черты лица и поведенческие расстройства. У подростков и взрослых лицо становится более угловатым, его черты огрубляются, выступающие лобные бугры и сросшиеся густые брови нередко придают лицу хмурый, насупленный вид. Половое созревание обычно проходит в нормальные сроки. Поведенческие расстройства с началом полового созревания, как правило, усиливаются, нарушения сна сохраняются. У взрослых усугубляется сколиоз. Агрессия, вспышки гнева, склонность к самоповреждению сохраняются, однако многие родители отмечают, что в зрелом возрасте больные становятся немного «спокойнее». Старейшей из ныне живущих больных сейчас около 85 лет.
Цитогенетическая диагностика
Примерно у 75% больных СМС имеется типичная делеция 17p11.2, соответствующая потере 4 миллионов нуклеотидов. У остальных больных отмечаются атипичные делеции, которые, затрагивая критический для СМС локус 17p11.2,выражаются в утрате большего или меньшего участка 17-й хромосомы. Все делеции 17p11.2, влекущие за собой СМС, включают делецию гена RAI1. Надежным методом диагностики СМС считается метод FISH, основанный на гибридизации хромосомного материала со специфическим для СМС флюоресцирующим ДНК_зондом, содержащим комплементарную RAI1 последовательность нуклеотидов. Этот метод позволяет выявить 95–100% делеций 17p11.2.
Распространенность
Распространенность СМС оценивают как 1 случай на 25 000 новорожденных, однако эта цифра представляется заниженной. Большинство случаев выявлено в течение последних 5–10 лет, что связано с развитием цитогенетической диагностики в мире. Синдром встречается повсеместно, во всех этнических группах. Почти все случаи СМС — спорадические. Данных в пользу влияния возраста родителей на частоту делеции нет.
По материалам сайта ассоциации «PRISMS»
Последние новости
Дата публикации: 07.04.2016
Новогодняя Ярмарка — 2019
Ежегодный праздник — Новогодняя Ярмарка в «особых мастерских») Технологического колледжа №21 состоится 19 декабря с 16.00 до 20.00
Дата публикации: 07.04.2016
Наше сообщество в Facebook
Дата публикации: 07.04.2016
Всемирный день осведомленности о синдроме Смит-Магенис (СМС)
17 ноября — Всемирный день осведомленности о синдроме Смит-Магенис (СМС)
Источник
Синдром Смита-Магениса (ССМ) может возникнуть у 1 из 25 000 детей. Но благодаря развитию цитогенетической диагностики он определяется все чаще. Встречается он спорадически и в любых этнических общностях.
Что такое ССМ?
Синдром Смита-Магениса — это патология, которая вызывает у ребенка задержку развития и определенные поведенческие и физические аномальные признаки.
Дети, пораженные этим синдромом, также могут страдать аутизмом, ОКР (обсессивно-компульсивным расстройством), СГДВ (синдромом гиперактивности и дефицита внимания) и расстройством настроения.
Помимо синдрома Смита-Магениса у таких детей часто встречаются наличие следующих медицинских проблем:
- полной или частичной потери слуха;
- почечных и сердечных дефектов;
- отслоения сетчатки глаза.
Симптоматика заболевания
Сопровождающие синдром Смита-Магениса симптомы проявляются в виде:
- особенных черт лица;
- нарушения умственной деятельности в разной степени проявления, ЗПР (задержка психического развития);
- регулярных изменений в поведении.
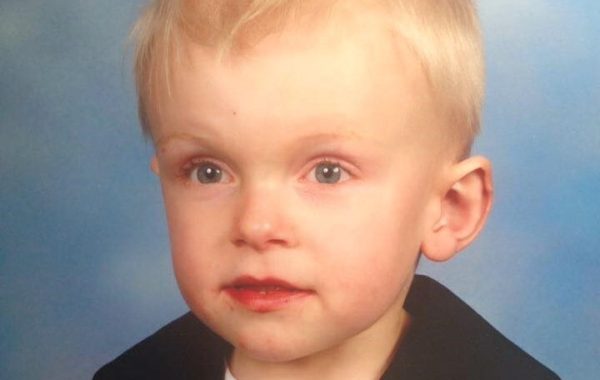
Часто внешность детей с ССМ схожа с внешностью детей с синдромом Дауна. Кроме этого, симптоматика физического характера у названных пациентов проявляется так:
- У них широкая форма лица с глубоко посаженными глазами. Сильно выступает вперед нижняя челюсть. Часто у таких детей диагностируется косоглазие и близорукость.
- У них обычно наблюдается пониженный тонус мышц, маленький рост, сколиоз и плоскостопие, хриплый голос, слабые рефлексы и низкая чувствительность боли.
- Пациенты страдают задержкой развития речи, нарушениями артикуляции.
- Для них характерны расстройства сна, то есть часто такие дети могут спать в дневные часы, а ночью бодрствовать. В младенчестве они могут не проснуться даже на кормление, приходиться их будить.
- Пациенты страдают приступами агрессии. Наступают они внезапно, и в эти моменты человек может нанести себе или другим людям серьезные увечья. Они царапаются, кусаются, ударяются о стену, выдергивают волосы.
Из других симптомов в поведении замечены склонности к объятиям самого себя, причем эти объятия довольно сильны.
Такие дети не могут сконцентрироваться надолго на одном занятии или предмете. Они часто либо очень импульсивны и гиперактивны, либо слишком вялые и сонные.
Как правило, родители таких детей отмечают, что они очень милые и добрые до тех пор, пока не наступают вспышки ярости. В такие моменты они сами могут пострадать от руки своего ребенка.
Причины синдрома Смита-Магениса
Причина, по которой возникает ССМ, заключается в том, что в гене 17-й хромосомы произошли изменения, присутствуют аномалии или не хватает небольшого участка.

Этот признак не всегда передается по наследству от родителей и поражает одинаково как мальчиков, так и девочек. Изменения, предшествующие появлению синдрома, происходят на стадии зачатия.
Лечение и помощь при ССМ
К сожалению, реалии сегодняшнего времени таковы, что лечению это заболевание не поддается. Нет никаких медицинских средств, при помощи которых можно было бы избавить новорожденного от синдрома.
Но родители в состоянии облегчить это положение и помочь малышу преодолеть самые затруднительные моменты. Если медицинское вмешательство осуществится на ранних сроках, то смогут помочь профессионалы. Это педагог, психолог, физиотерапевт, логопед и др.

Важно, чтобы родители задавали как можно больше вопросов специалистам. Ведь чем больше известно информации о проблеме, тем больше возможностей ее решить.
Кроме того, родители могут связаться с другими людьми, воспитывающих таких детей, а также воспользоваться услугами психолога в личных целях. Так как для ухода за такими детьми сами родители должны быть здоровы физически и умственно.
Источник
Русский Медицинский Сервер / Лечение в Италии / Центр лечения редких заболеваний в Милане / Синдром Смит-Магенис — лечение в Италии
Синдром Смит—Магенис (Smith—Magenis syndrome)—это генетическое нарушение, возникающее при отсутствии небольшого участка 17-й хромосомы [обозначается как del(17р11.2)] и проявляющееся характерным строением тела, особенностями развития и поведения. Первая группа детей была описана в 1980 году в США врачом-клиницистом Энн Смит и цитогенетиком Эллен Магенис.
Симптомы:
- характерные черты лица (широкое квадратное лицо; короткоголовость, то есть относительно большие поперечные размеры головы; выпуклый лоб; сросшиеся брови; монголоидный разрез глаз; глубоко посаженные глаза; широкая переносица; недоразвитие средней трети лица; короткий нос с открытыми вперед ноздрями; малая высота носа; малые размеры нижней челюсти, а со временем — выступание нижней челюсти вперед; вывернутая наружу мясистая верхняя губа, имеющая форму шатра);
- низкий рост, маленькие кисти и стопы, короткие пальцы рук и ног, плоскостопие, необычная походка (с широко расставленными ногами);
- задержка физического развития;
- низкий мышечный тонус;
- проблемы вскармливания (выраженная орально-моторная дисфункция с нарушениями сосания и глотания, со слабостью мышц языка, с вытеканием слюны изо рта; отказ от пищи определенной консистенции, желудочно-пищеводный рефлюкс);
- низкий хриплый голос;
- хронический средний отит, тугоухость;
- затяжные риниты и синуситы, требующие лечения антибиотиками;
- косоглазие, близорукость и другая патология глаз;
- ослабленные рефлексы, признаки периферической нейропатии, пониженная чувствительность к боли;
- частые запоры;
- сколиоз;
- повышенный уровень холестерина в крови;
- задержка психического развития;
- умственная отсталость (легкая или средней тяжести);
- задержка речевого развития, проблемы с артикуляцией;
- дефицит внимания, часто в сочетании с гиперактивностью;
- нарушения сна (ночной сон менее глубокий, с частыми пробуждениями; ранние утренние подъемы; дневная сонливость), обусловленные неправильным суточным ритмом секреции мелатонина;
- стереотипное поведение: порывистое сжатие руками верхней половины тела (самообъятие); перелистывание страниц, слюнявя палец; стремление все засовывать в рот, в том числе собственные руки; скрежетание зубами;раскачивание туловища; верчение и кручение предметов;
- дезадаптивное поведение: частые вспышки гнева, требование внимания к себе со стороны взрослых, импульсивность, отвлекаемость, непослушание, агрессия, проблемы с отправлением естественных потребностей;
- склонность к самоповреждению (больные могут сами себя бить, кусать, щипать, вводить инородные тела в естественные отверстия тела, сдирать ногти на руках и ногах);
- реже встречаются патология сердца, почек и мочевых путей, нарушения функции щитовидной железы, эпилепсия, нарушения иммунитета, расщелина неба и расщелина губы, отслойка сетчатки.
В детстве этот синдром проявляется повышенной сонливостью – ребенок самостоятельно не просыпается, для кормления, его необходимо разбудить. Такие дети склоны наносить себе различные увечья. Лица, страдающие синдромом Смита-Магениса склонны сами себя обнимать, причем порой эти объятия характеризуются большой силой. Однако несмотря на достаточную специфичность клинической картины, диагноз этого синдрома устанавливается на основании результатов генетического исследования.
Данное исследование проводится в семье, состоящей из трех человек (папа, мама и ребенок). Это даст возможность определить носителей, которые не имеют клинических проявлений. Например, либо мать, либо отец являются носителями мутантного гена, но не все дети могут быть больными. В последующем с помощью медико-генетического консультирования появится возможность рождения только здоровых детей.
При этом дети обычно ласковы, добродушны, общительны, любят внимание и поощрение, эмоциональны, легко подражают другим, любят музыку, обладают чувством юмора и хорошей долговременной памятью на имена, места и события.
Диагноз нередко ставится только в школьном возрасте, когда становятся очевидными характерные черты лица и поведенческие расстройства. У подростков и взрослых лицо становится более угловатым, его черты огрубляются, выступающие лобные бугры и сросшиеся густые брови нередко придают лицу хмурый, насупленный вид. Половое созревание обычно проходит в нормальные сроки.
Поведенческие расстройства с началом полового созревания, как правило, усиливаются, нарушения сна сохраняются. У взрослых усугубляется сколиоз. Агрессия, вспышки гнева, склонность к самоповреждению сохраняются, однако многие родители отмечают, что в зрелом возрасте больные становятся немного «спокойнее». Старейшей из ныне живущих больных сейчас около 85 лет.
Распространенность СМС оценивают как 1 случай на 25 000 новорожденных, однако эта цифра представляется заниженной. Большинство случаев выявлено в течение последних 5–10 лет, что связано с развитием цитогенетической диагностики в мире. Синдром встречается повсеместно, во всех этнических группах. Почти все случаи СМС — спорадические. Данных в пользу влияния возраста родителей на частоту делеции нет.
Выявление участка хромосомы, в котором отсутствует определенный фрагмент, свойственный синдрома Смит-Магенис, позволяет выставить данный диагноз. В зависимости от конкретного диагноза проводится соответствующее лечение. В ведении таких пациентов очень большая роль отводится реабилитационным мероприятиям, которые включают в себя логопедическую, моторную помощь и т.д. Для борьбы с повышенной сонливостью назначаются определенные лекарственные препараты. Повышенная двигательная активность и психомоторное возбуждение являются показаниями для использования психотропных лекарственных средств.
! Несмотря на то, что многие из описанных в данном разделе болезней считаются неизлечимыми, в Центре лечения редких заболеваний в Милане постоянно ведется поиск новых методов. Благодаря генной терапии удалось добиться выдающихся результатов и полностью излечить некоторые редкие синдромы.
Обратитесь к консультанту на сайте или оставьте заявку — так вы можете узнать, какие методы предлагают итальянские врачи. Возможно, данное заболевание уже научились лечить в Милане.
+7 (925) 50 254 50 –
срочное лечение в Италии
ЗАПРОС в КЛИНИКУ
Источник