Что такое синдром нагера фото

Среди генетических заболеваний одним из самых редких признана патология, которую впервые в середине прошлого века описали учёные из Швейцарии Рейнер и Нагер. В честь второго заболевание и было названо. С тех пор синдром Нагера постоянно изучают врачи всех стран, стараясь найти способ предотвратить появление болезни или помочь больному жить, не страдая от собственной неполноценности. Сегодня врачи способны поставить точный диагноз, выявив болезнь на ранних сроках беременности. С помощью ультразвукового исследования медики получают достоверную информацию о состоянии костей и слухового прохода плода, находящегося в утробе матери.
Особенности недуга
Заболевание, которое описали учёные в конце 40-х годов ХX столетия, встречается редко, но каждый отдельный случай заслуживает внимания медиков. Давно доказано, что синдром Нагера – результат генной мутации, справиться с которой почти невозможно, но врачи обнаруживают признаки патологии при первом посещении беременной женщиной кабинета УЗИ.
С помощью ультразвука обнаруживают:
- Недостаточный уровень развития костей нижней челюсти плода или полное отсутствие челюстных сочленений.
- Недоразвитие или отсутствие костей, идущих от запястья к пальцам.
- Изменения в формировании костей пальцев рук (6 и более пальцев или их отсутствие).
- Неправильную (аномальную) форму разреза глаз (опущение верхнего или нижнего века).
- Нарушения строения и формирования слухового аппарата.
Если у ребёнка, появившегося на свет, сразу после рождения обнаруживается болезнь, которую описал Нагер, значит, малыш будет абсолютно глухим, а челюстная аномалия станет причиной нарушенной артикуляции. Однако у детей с таким диагнозом нормальный интеллект, несмотря на задержку в психомоторном развитии.
Количество отклонений от нормы в формировании различных органов и систем детского организма не одинаково у всех пациентов.
 Проявления патологии проявляются в виде аномальных отклонений от нормы:
Проявления патологии проявляются в виде аномальных отклонений от нормы:
- Гидроцефалия и сколиоз.
- Микроцефалия и недоразвитие шейного отдела позвоночника.
- Нарушение в развитии полушарий головного мозга.
- Стеноз ликворного русла.
- Порок сердца и аномальное строение кровеносных сосудов.
Встречаются случаи, когда малыш появляется на свет с пороками развития не совместимыми с жизнью, но в основном новорождённые выживают и живут на протяжении долгого времени.
Причины возникновения патологии и её симптоматика
В чём причина генетического сбоя, учёные не знают и сегодня. Однако доподлинно известно, что с синдромом, который описал Nager, рождаются дети не у всех родителей, имеющих мутировавший ген.Порой даже если отец и мать носители такого гена, их малыш может родиться абсолютно здоровым, а у здоровых мужчин и женщин рождаются больные дети. Причина – спонтанная мутация:
- Это значит, что будущая мать подверглась радиоактивному облучению во время беременности или жила в населённом пункте, расположенном неподалёку от места разработки и добычи радиоактивных руд.
- Не менее опасно для будущих младенцев увлечение их родителей наркотическими веществами или алкоголем.
- Токсикомания приводит к видоизменению генов и спонтанной мутации. Результат – рождение малыша с синдромом Нагера.
 Как утверждают учёные-медики, синдром Нагера диагностируется у половины детей, получивших мутировавший ген от обоих родителей. Вторая половина таких новорождённых – абсолютно здоровые дети.
Как утверждают учёные-медики, синдром Нагера диагностируется у половины детей, получивших мутировавший ген от обоих родителей. Вторая половина таких новорождённых – абсолютно здоровые дети.
Риск произвести на свет больного малыша слишком велик, поэтому все женщины, ставшие на учёт в женской консультации, стремятся пройти обследование и получить ответ на свой вопрос.
Результаты УЗИ позволят принять правильное решение, спланировать ведение беременности и принять решение относительно способа родоразрешения.
Характерный симптом акрофациального дизостоза (синдрома Нагера) – аномалии в развитии нижней челюсти. Этот челюстно-лицевой дизостоз сочетается с другими признаками заболевания:
- Недоразвитие или отсутствие луча первого пальца кистей рук (мизинцев). В некоторых случаях отсутствует или недостаточно развит луч безымянного пальчика.
- Недостаточное развитие лучевых костей и, соответственно, неестественное положение пальцев рук.
- Укорочение твёрдого нёба.
- Недоразвитие коренных зубов.
- Неправильная форма ушных раковин.
- Антимонголоидный разрез глаз (опущение верхнего или нижнего века).
У детей, рождённых с синдромом Нагера, могут быть искривлены нижние конечности или проявляется дизостоз на других участках тела (недоразвиты и деформированы ключицы, лопатки, скулы).
Чего ждать в будущем
 Прогноз, о котором говорят врачи, не столь печален. Существуют определённые методы лечения, применяемые при наличии незначительных аномалий в развитии ребёнка. В большинстве случаев проводится операция по устранению деформаций, в ходе которой:
Прогноз, о котором говорят врачи, не столь печален. Существуют определённые методы лечения, применяемые при наличии незначительных аномалий в развитии ребёнка. В большинстве случаев проводится операция по устранению деформаций, в ходе которой:
- Восстанавливается форма кистей и пальцев рук (разделение сросшихся фаланг).
- Коррекция формы ушных раковин.
- Восстановление внутренних органов.
Если диагноз был поставлен на ранних сроках после зачатия, родителям предлагают согласиться на искусственное прерывание беременности (аборт).
Существуют и более сложные техники хирургического вмешательства, применяемые очень редко. Современные акушеры-гинекологи в некоторых случаях с помощью кесарева сечения частично извлекают плод из полости матки, проводят трахеотомию, обеспечивая крохе возможность самостоятельно дышать, затем операция завершается.
В других случаях малышу хирургическим путём формируют слуховой проход или вживляют специальный аппарат. С его помощью ребёнок слышит. Это вмешательство называется бинауральное слухопротезирование. Во время операции врачи используют имплантируемые цифровые слуховые аппараты, обеспечивающие качественное восприятие звуков.
 Большую работу проводят стоматологи и хирурги, проводящие челюстно-лицевые операции. Их цель – восстановление формы нижней и верхней челюсти, зубного ряда. С помощью данного вмешательства маленькому пациенту обеспечивают возможность не только разговаривать, но и пережёвывать пищу. Деятельность ортопедов направлена на устранение деформаций верхних и нижних конечностей, восстановление их функциональности и качественной подвижности.
Большую работу проводят стоматологи и хирурги, проводящие челюстно-лицевые операции. Их цель – восстановление формы нижней и верхней челюсти, зубного ряда. С помощью данного вмешательства маленькому пациенту обеспечивают возможность не только разговаривать, но и пережёвывать пищу. Деятельность ортопедов направлена на устранение деформаций верхних и нижних конечностей, восстановление их функциональности и качественной подвижности.
Продолжительность жизни пациентов, рождённых с диагнозом «акрофациальный дизостоз» (синдром Нагера) составляет от 20 до 50 лет. Зависит она от состояния общего здоровья, степени функциональности внутренних органов и тяжести заболевания.
Учитывая отсутствие нарушений в психомоторном развитии, можно смело сказать, что пациенты с синдромом Нагера способны вести активную жизнь, несмотря на необходимость создания специальных условий для некоторых из них.
Чаще всего жизненные трудности связаны с нарушением слуха, но регулярные занятия с сурдопедагогом помогают пациентам наладить контакт с окружающими. Своевременно проведённая операция по вживлению слухового аппарата позволит малышу избежать отставания в речевом и психическом развитии.
Сложность представляют случаи, когда врачам приходится сталкиваться с самой тяжёлой 4 степенью нарушения формирования и развития лучевой кости, гортани и надгортанника, а также с:
- отсутствием оболочки органов зрения;
- полным сращением пальцев стопы;
- укорочением конечностей.
Потребуется масса усилий и длительное лечение, чтобы помочь пациенту стать полноценным членом общества. Современные пластические и челюстно-лицевые хирурги, ортопеды, стоматологи и другие специалисты способны устранить большую часть дефектов в развитии малыша.
Проводя сложные операции, медики добиваются восстановления функциональности не только конечностей, но и внутренних органов. Хирургическое вмешательство проводится детишкам с синдромом Нагера сразу после появления на свет. Это даёт им возможность свободно дышать и глотать. Отсутствие отставания в других сферах развития обеспечивает таким пациентам нормальную продолжительность жизни.
Чтобы уберечь будущего ребёнка от столь серьёзного заболевания, нужно заранее пройти обследование, получить консультацию опытного генетика, вести здоровый образ жизни, заботясь о собственном здоровье и о здоровье долгожданного малыша.
Источник
Синдром Тричера Коллинза был впервые описан британским врачом Томпсоном в 1846 г. На фото пациентов отчетливо прослеживаются одни и те же признаки аномалий лицевых костей. По мере роста ребенка они становятся более выраженными. Наилучший прогноз жизни у тех пациентов, у которых не нарушено развитие внутренних органов.
Общая информация
Синдром Тричера Коллинза, фото которого можно увидеть в этой статье, – это генетическое заболевание, при котором происходит мутация гена 5-й хромосомы, отвечающего за формирование костей лицевой части черепа. Кроме этого, могут быть и другие нарушения.
При данном заболевании прекращается нормальный процесс синтеза и удвоения ДНК. Это происходит на 3-4 неделе после оплодотворения. В результате уже на втором месяце беременности в костях лица формируются участки из соединительной ткани, так как половины генетического материала становится недостаточно для нормального развития организма.
Деление клеток в структурах правой жаберной дуги зародыша нарушается.
Болезнь передается по наследству и может наблюдаться у членов одной семьи в 2-3 поколениях. Она имеет различный характер и степень тяжести, что затрудняет генетическое консультирование (профилактику наследственных болезней). Распространенность составляет 1 случай на 50 тыс. новорожденных.
Измененная копия гена является доминирующей, поэтому риск рождения ребенка с этой патологией высок, даже если болен только один из родителей (около 90% вероятности). В случае, когда данным заболеванием страдают оба родителя, то такая вероятность составляет 100%.
В медицине существуют и другие названия синдрома Тричера Коллинза:
- синдром Франческетти;
- мандибулофациальный дизостоз;
- синдром Томпсона.
Почему появляется заболевание
Несмотря на то, что данная патология наследуется от больных родителей, в 60% случаях происходит первичное развитие новых мутаций, которые вызваны следующими негативными факторами:
Их влияние на организм женщины до беременности или во время нее может вызвать возникновение дефекта в генах.
Симптомы
Синдром Тричера Коллинза, фото которого наглядно демонстрирует характерные особенности заболевания, обладает следующими основными признаками:
- антимонголоидный разрез глаз (89% пациентов), при котором их наружный угол опущен вниз;
- недоразвитие или искажение формы нижней челюсти, скуловых костей и орбит глаз (80% случаев), имеющее двусторонний, симметричный характер;
- отсутствие части тканей века (чаще всего в виде треугольной вырезки) или ресниц на нижнем веке;
- «птичье лицо», формирующееся из-за недоразвития нижней челюсти (78% детей), западения подбородка назад и относительно крупного носа;
- «затонувшее лицо» – форма, при которой его центральная часть находится как бы в углублении по сравнению с нормой;

- узкие ноздри;
- плоский угол между носом и лбом (у здоровых людей он равен 115-130°);
- недоразвитие ушных раковин, их грубая деформация или полное отсутствие;
- открытый прикус, при котором отсутствует смыкание верхней и нижней челюстей;
- неправильный рост зубов во рту, их недоразвитие (особенно моляров), широкое расстояние между ними;
- готическое (высокое) небо, расщелина в нем (28% больных);
- «волчья пасть», при которой в результате образования расщелины ротовая и носовая полость соединяются между собой.
2 последних признака проявляются реже.
Сопутствующие проблемы
Кроме вышеописанных симптомов, могут наблюдаться следующие нарушения:
- аномалии костной системы, в том числе позвоночника;
- отсутствие околоушных слюнных желез;
- врожденные пороки сердца;
- доброкачественные новообразования из хрящевой ткани внутри костей;
- околоушные свищи;
- неопущение яичек в мошонку у мальчиков;
- недоразвитие гайморовых пазух;
- нарушения в кровеносной системе – анемия, нейтропения (снижение концентрации гранулоцитов в крови, приводящее к ухудшению иммунитета), гемолитическая желтуха в результате повышенного разрушения красных кровяных клеток;
- умственная отсталость;
- повышенное оволосение на отдельных участках тела.
Дыхание
У многих новорожденных фиксируется сужение дыхательных путей. Ограниченная возможность для открытия рта и выпадающий язык могут привести к трудностям дыхания и питания в раннем возрасте.
Часто также присутствуют следующие нарушения:
- хроническая дыхательная недостаточность, которая сопровождается одышкой, тахикардией, проявлениями со стороны нервной системы;
- апноэ во время сна – задержка легочной вентиляции более чем на 10 сек., вызывающее состояние гипоксии, а в некоторых случаях – легочную гипертензию с недостаточностью правого желудочка сердца;
- атрезия хоан – аномальное развитие внутренних носовых ходов, при котором происходит их заполнение соединительной тканью.
Слух
Кроме видимых нарушений в наружном аппарате уха, у больных также имеются следующие отклонения:
- двусторонняя тугоухость, возникающая в результате ухудшения передачи звуковых волн от наружного к внутреннему уху (снижение слуха на 50-70 дБ);
- патологическое строение среднего уха, иногда его полное отсутствие;
- полное или частичное отсутствие слуховой трубы;
- пороки развития внутреннего уха (в редких случаях);
- деформация слуховых косточек.
Зрение
Синдром Тричера Коллинза, для которого характерен специфический разрез глаз, показанный на фото, сопровождается также следующими нарушениями органов зрения:
- недоразвитие глазного яблока;
- врожденная катаракта (помутнение хрусталика, вызывающее снижение зрения);
- кисты по краю роговицы и на конъюнктиве;
- расщепление сосудистой оболочки глаза и зрительного нерва;
- недоразвитость, слабость (или отсутствие) глазодвигательных мышц;
- отсутствие мейбомиевых желез, участвующих в образовании слезной жидкости (чаще на нижнем веке).
Руки
На кистях рук и стопах у больных данным синдромом в некоторых случаях отмечается недоразвитие ногтевых пластин.
Диагностика во время беременности
Диагностика заболевания плода в период его вынашивания производится 2 способами: с помощью ультразвукового исследования, на котором выявляются грубые челюстно-лицевые нарушения, и молекулярного исследования образцов биологического материала.
Молекулярно-генетическая диагностика позволяет выявить изменения в ответственном гене путем прямого определения аминокислотной последовательности, которое производится автоматически.
В качестве биологического материала используются:
- образцы ворсин внешней оболочки зародыша, если срок беременности находится в пределах 8-14 недель;
- околоплодные воды, если женщина беременна на сроке 16-21 недель.
Стоимость исследования составляет порядка 30 тыс. руб., срок выполнения около 1 месяца, цена поиска мутации у родственника – 2800-3500 тыс. руб.
Если в семье уже имелись подобные случаи генетических нарушений, то настоятельно рекомендуется сделать диагностику перед подготовкой к беременности (стоимость исследования – 8-10 тыс. руб.). Такое исследование является единственным способом профилактики возникновения данной патологии у ребенка.
Диагностика у новорождённого
Определение заболевания у новорожденного обычно не вызывает затруднений и производится по внешним признакам. Генетический анализ для дополнительного подтверждения диагноза производится по венозной крови, таким же способом, как во время беременности.
Стоимость такого анализа порядка 60 тыс. руб. (вместе с заключением врача-генетика). При выявлении отклонения также рекомендуется сделать исследование для обоих родителей, братьев и сестер.
Дополнительно проводятся следующие обследования:
- определение остроты слуха, методика проведения зависит от возраста ребенка: регистрация вызванных слуховых потенциалов (подача акустических сигналов и регистрация электроэнцефалограммы), речевая аудиометрия (произнесение слов и фраз с различной громкостью), тональная пороговая аудиометрия (проводится с помощью аудиометров, результатом является графическое изображение слухового порога);
- рентгенография черепа;
- компьютерная томография височных костей по достижении возраста 3 лет для подготовки к хирургической операции;
- консультация у профильных специалистов (отоларинголога, окулиста, кардиолога, хирурга и других).
Данное заболевание требует дифференциальной диагностики с такими генетическими патологиями, как:
- Синдром Нагера. Для этой болезни также характерен антимонголоидный разрез глаз, недоразвитие нижней челюсти и патологические изменения в органах слуха и зрения. Кроме этого, у больных детей имеются грубые нарушения в развитии конечностей – укорочение и искривление предплечья, фаланг и локтевых костей, отсутствие пальцев и сращение костей рук.

- Синдром Гольденхара. Неправильное развитие затрагивает не только лицо и череп, но и нервную, сердечно-сосудистую, мочевыделительную, пищеварительную системы, у многих больных отмечается умственная отсталость. Как правило, данная аномалия имеет односторонний характер (70% больных).
Стадии развития болезни
Синдром Тричера Коллинза, фото которого приведено выше, условно делят на 3 стадии:
- Начальный этап, когда у новорожденного наблюдается незначительное деформирование лицевой части черепа.
- 2-я стадия, на которой происходят нарушение проходимости дыхательных путей, глотания, зрения и слуха.
- 3-я стадия, которая характеризуется грубыми деформациями черепа из-за неравномерного роста костей в течение многих лет.
Первые признаки заболевания можно заметить сразу после рождения ребенка. Их выраженность может быть различной – от едва заметных отклонений до очень тяжелых форм. Анатомические аномалии без хирургического вмешательства постепенно прогрессируют по мере роста и развития ребенка.
Как живут дети с синдромом
В медицинской литературе описано около 250 случаев данного заболевания. И некоторые из них получили мировую огласку.
Так, в 2013 г. вышла передача об американской девочке Джулианне Уэтмор, у которой при рождении была диагностирована самая тяжелая степень этого заболевания в истории медицины. У нее отсутствовало 30-40% костей лица. В прессе ее называли девочкой, родившейся без лица.
 Джулианне Уэтмор с синдромом Тричера Коллинза
Джулианне Уэтмор с синдромом Тричера Коллинза
Несмотря на это, она имеет нормальный уровень интеллекта и является жизнерадостным, общительным ребенком. Ее история стала вирусной через год после рождения в 2003 г. Девочка перенесла несколько десятков пластических операций и теперь ее лицо выглядит гораздо лучше, а ее показательный случай дает надежду другим родителям, столкнувшимся с такой же проблемой.
Семья Джулианны усыновила еще одну девочку с таким же генетическим заболеванием. Первая дочь родилась у них совершенно здоровой, однако, зная о том, что патология передается по наследству, они отказались от идеи рождения 3-го ребенка.
Хирурги надеются, что девочка сможет посещать колледж. Из-за того, что ее челюстные кости сильно деформированы, ей приходится питаться через зонд. Есть сложности и с речью, но она быстро учится языку жестов.
Обучение таких детей чаще всего происходит в специализированных школах для больных с нарушениями слуха. Способность к социальной адаптации во многом зависит от родителей ребенка и выполнения рекомендаций врачей.
Возможные осложнения и прогноз
Если заболевание не сопровождается грубыми пороками развития внутренних органов, то прогноз для жизни и здоровья ребенка в будущем благоприятен. При наличии тугоухости снижается способность к развитию речи, письма, обучению.
Необычная внешность также мешает получению навыков социального общения, так как другие дети стараются избегать сверстников с врожденными аномалиями и уродствами. В результате у ребенка может быть значительно снижена самооценка, что требует консультации со стороны психолога. Проведение ряда пластических операций позволяет частично сгладить дефекты развития.
Тугоухость часто ошибочно диагностируется как недоразвитие психики. Умственная отсталость у таких детей встречается редко, а некоторые имеют способности выше среднего. Поэтому у таких пациентов важно на ранней стадии выявлять нарушения слуха и корректировать их.
Врачи рекомендуют это делать в обязательном порядке до достижения ребенком шестимесячного возраста. С 3-х месяцев он может носить аппарат для костного проведения звуковых волн, а после 3 лет возможна установка имплантата за ушами. Также очень большое значение имеют занятия с сурдопедагогом.
В первые месяцы жизни недоразвитость нижней челюсти может способствовать выпадению языка и перекрытию дыхательных путей, что является потенциально опасным для жизни ребенка. В последующем могут возникнуть затруднения в приеме пищи из-за ограниченной возможности открытия рта (в различной степени тяжести).
Продолжительность жизни
Синдром Тричера Коллинза, фото которого свидетельствуют об челюстно-лицевых аномалиях, в легких случаях практически не влияет на продолжительность жизни. Гибель пациентов с данным заболеванием наступает при присоединении других патологий, осложняющих его течение. Многие из них доживают до зрелого возраста и даже заводят семьи.
Однако в тяжелых случаях, при множественных аномалиях органов и систем, генетический дефект дает летальный исход еще во внутриутробный период или в первые дни после рождения ребенка.
Об этом свидетельствует высокая частота выкидышей и ранних детских смертей. Врачи отмечают также, что при передаче заболевания к следующему поколению имеется тенденция к увеличению тяжести и осложнений в том случае, если носителем дефектного гена является мать.
Лечебные мероприятия
Как и для большинства генетических заболеваний, специального лечения при данном синдроме не существует. Терапия представляет собой сложную, многопрофильную задачу и зависит от тяжести поражения и осложнений.
Проводятся следующие лечебные мероприятия:
- В случае дыхательной недостаточности – трахеостомия. Эта процедура заключается в создании проходимости в трахее с помощью установки трубки для обеспечения поступления воздуха в дыхательные пути. В легких случаях возможна неинвазивная искусственная вентиляция легких.
- Дистракция (растяжение) нижней челюсти хирургическим путем с помощью специального аппарата. Необходимость проведения такой манипуляции рассматривается врачами в индивидуальном порядке.
- Установка гастростомы – трубки, которая требуется для защиты дыхательных путей от попадания пищи.
- Хирургическая реконструкция лицевой части черепа. Она обычно проводится с 5-6 лет.
При наличии осложнений показаны консультации у соответствующих специалистов и патогенетическое лечение.
Хирургические способы восстановления слуха у таких детей неэффективны. Рекомендуемая тактика реабилитации – использование слуховых аппаратов костной проводимости (или обычных слуховых аппаратов при незначительной деформации ушной раковины).
Их особенностью является то, что звуки передаются во внутреннее ухо по костям черепа. Это не так физиологично, как воздушное звукопроведение, однако при определенном усилении они очень хорошо воспринимаются рецепторами.
При данном заболевании хорошо зарекомендовали себя имплантируемые слуховые аппараты ВАНА (БАХА), которые имеют в своем составе титановую опору, закрепляемую в толще височной кости. Со временем титан срастается с костной тканью, а штифт напрямую передает звуковые колебания в улитку внутреннего уха.
Хирургическая установка имплантата производится в 2 этапа: сначала внедряется титановый штифт, а затем, после его вживления в кость в течение полугода, монтируется опора. Через месяц на нее надевают звуковой процессор.
Такие слуховые аппараты обладают следующими преимуществами:
- у детей отмечается улучшение порогов слышимости в диапазоне громкости обычной речи;
- по сравнению с обычными аппаратами и хирургической реконструкцией эстетические и аудиологические показатели выше;
- происходит спонтанное улучшение речи и постановки голоса (его высоты и интенсивности).
У маленьких детей слухопротезирование проводится с применением эластичной ленты, закрепляемой на голове. Существуют также цифровые аппараты костного проведения звуков, имплантируемые в височную кость (Alpha).
Операция
Аномальное развитие лицевых костей помогает устранить челюстно-лицевая и пластическая хирургия. В сложных случаях требуется проведение целой серии таких операций.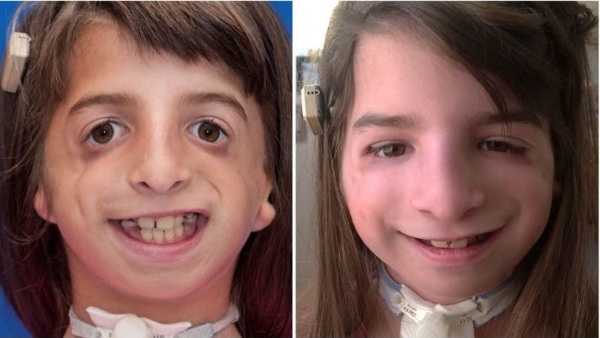
Оперативной корректировке поддаются следующие нарушения:
- дефекты ушной раковины и среднего уха;
- искаженный овал лица (липоскульптура, вытяжение, костные трансплантаты);
- «волчья пасть»;
- расщелины неба;
- дефекты век;
- ограниченное открытие рта.
Этиологического лечения для синдрома Тричера Коллинза не существует. Как показывают фото, степень выраженности этого генетического отклонения может быть различной. Для пациентов применяется терапия, поддерживающая их жизнь на достаточном уровне.
Наиболее распространенным осложнением является ухудшение слуха, для устранения которого используются слуховые аппараты. С целью восстановления анатомических дефектов по возможности проводят пластическую хирургию.
Оформление статьи: Владимир Великий
Видео о синдроме Тричера Коллинза
Девочка с синдромом Тричера Коллинза:
Источник