Что такое синдром марото лами

Синдром Марото — Лами (мукополисахаридоз VI типа[1], англ. MPS VI) — редкая наследственная болезнь, одна из форм мукополисахаридоза из группы лизосомных болезней накопления, биохимически связанная с дефицитом фермента лизосом N-Ацетилгексозамин-4-сульфатсульфатазы (арилсульфатазы Б, англ. arylsulfatase B, ARSB)[2].
Историческая справка[править | править код]
Эпоним[править | править код]
Данный тип мукополисахаридоза получил название в честь французских врачей: педиатра Пьера Марото (фр. P. Maroteaux, род. в 1926) и его наставника генетика Мориса Эмиль Жозеф Лами (фр. M. E. J. Lamy, 1895—1975)[3][4].
Патогенез[править | править код]
В результате мутации гена 5q11-13 (ARSB) развивается дефицит или проявляется дефектность фермента N-Ацетилгексозамин-4-сульфатсульфатазы, что ведёт к накоплению в лизосомах одного типа гликозаминогликанов — дерматансульфата[5][6].
Наследование[править | править код]
Аутосомно-рецессивный механизм наследования синдрома Марото — Лами: оба родителя являются носителями дефектного гена (помечен красным кружочком). По законам Менделя 50 % детей станут носителями (как их родители), 25 % родятся генетически здоровыми и в 25 % случаев — больными.
Данное генетическое заболевание наследуется, как и подавляющее большинство лизосомных болезней накопления, по аутосомно-рецессивному типу наследования. Таким образом, с одинаковой частотой встречается как у мужчин, так и у женщин.
Аутосомно-рецессивный тип наследования на практике означает, что дефектный ген расположен на одной из двух аллельных аутосом. Заболевание клинически манифестирует только в случае, когда обе аутосомы, полученные по одной от отца и матери, являются дефектными по данному гену. Как и во всех случаях аутосомно-рецессивного наследования, если оба родителя несут дефектный ген, то вероятность наследования болезни у потомства составляет 1 из 4. Таким образом в среднем, на одного больного ребёнка в такой семье приходится три без клинических признаков проявлений генной болезни. На схеме синим цветом обозначены здоровые, фиолетовым — носители дефектного гена, красным — синдром Марото — Лами (два дефектных гена одной аллели). Синим кружочком помечен нормальный ген, красным — дефектный.
Классификация[править | править код]
Согласно Международной классификации болезней десятого пересмотра (МКБ-10), различают:
- E7676. Нарушения обмена гликозаминогликанов
- E76.276.2 Другие мукополисахаридозы. Недостаточность β-глюкуронидазы. Мукополисахаридозы типов III, IV, VI, VII. Синдром: Марото — Лами (лёгкий), (тяжёлый), Моркио(-подобный), (классический), Санфилиппо (тип A) (тип В) (тип С) (тип D).
Клиническая картина[править | править код]
Интеллектуальное развитие детей с синдром Марото — Лами, как правило, не страдает (нормальное)[5], тем не менее, наблюдается множество общих черт с синдромом Гурлер. Вызываемый дефицитом фермента N-Ацетилгексозамин-4-сульфатсульфатазы синдром Марото — Лами обладает разнообразным спектром серьёзных клинических симптомов. Неврологические осложнения включают помутнение роговицы, развитие глухоты, утолщение твёрдой мозговой оболочки (одной из трёх мембран, окружающих и защищающих головной и спинной мозг), болевой синдром, вызванный сжатием или травмированием нервных корешков и периферических нервных волокон. Первые признаки болезни проявляются на первом году жизни ребёнка — одним из первых симптомов зачастую является отставание в моторном развитии (дети позже начинают ходить)[7]. В возрасте 10 лет у детей наблюдается укорочение туловища, своеобразная поза «на корточках», вызванная ограничением подвижности суставов. В более тяжёлых случаях у детей развивается характерный выпирающий живот, возникающий в результате избыточного искривления вперёд поясничного отдела позвоночного столба (гиперлордоз). Скелетные деформации прогрессируют (особенно в области таза), способствуя дальнейшему ограничению объёма движений в суставах. У многих детей формируется пупочная или паховая грыжа. Практически у всех детей встречаются различные формы заболеваний сердца, как правило, проявляющиеся дисфункцией клапанов.
Диагностика[править | править код]
Лечение[править | править код]
Современная наука получила возможность проводить фермент-заместительную терапию для пациентов с мукополисахаридозом ферментом лизосом, участвующим в катаболизме мукополисахаридов, составляющих основу межклеточного вещества соединительной ткани. У больных синдромом Марото — Лами, фермент-заместительная терапия оказалась относительно успешной: улучшились развитие и подвижность суставов. В дальнейшем предпринимались попытки инъекции недостающего фермента в бёдра, с целью увеличения объёма движений и купирования болевого синдрома. Тем не менее, при стоимости терапии $ 365 000 в год — лечение является одним из самых дорогих[8].
См. также[править | править код]
- Лизосомные болезни накопления
- Мукополисахаридоз
Примечания[править | править код]
- ↑ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. Dermatology: 2-Volume Set (неопр.). — St. Louis: Mosby, 2007. — ISBN 1-4160-2999-0. (англ.)
- ↑ Garrido E., Cormand B., Hopwood J.J., Chabás A., Grinberg D., Vilageliu L. Maroteaux-Lamy syndrome: functional characterization of pathogenic mutations and polymorphisms in the arylsulfatase B gene (англ.) // Mol. Genet. Metab. (англ.)русск. : journal. — 2008. — July (vol. 94, no. 3). — P. 305—312. — doi:10.1016/j.ymgme.2008.02.012. — PMID 18406185. (англ.)
- ↑ synd/1619 на Who Named It? (англ.)
- ↑ Maroteaux P., Leveque B., Marie J., Lamy M. A new dysostosis with urinary elimination of chondroitin sulfate B (фр.) // Presse Med (англ.)русск.. — 1963. — Сентябрь (т. 71). — С. 1849—1852. — PMID 14091597.
- ↑ 1 2 Т. Р. Харрисон. Внутренние болезни в 10 книгах. Книга 8. Пер. с англ. М., Медицина, 1996, 320 с.: ил. Глава 316. Лизосомные болезни накопления (с. 250—273). med-books.info. Дата обращения 14 декабря 2014.
- ↑ Справочник Т. Р. Харрисона по внутренним болезням, 1992—1997:. Глава 316. Лизосомные болезни накопления. rusmedserver.ru. Дата обращения 14 декабря 2014.
- ↑ Topic Galleries, Chicago Tribune. (недоступная ссылка) (англ.)
- ↑ Health Care: The World’s Most Expensive Drugs, Matthew Herper, Forbes, Feb. 22, 2010 (англ.)
Ссылки[править | править код]
- Maroteaux-lamy.com (англ.)
- Harrison’s Principles of Internal Medicine.
Источник
Мукополисахаридоз VI типа относится к редким (орфанным) заболеваниям. Частота его оценивается как 1:300 000 живорожденных. На сегодняшний день в мире насчитывается около 1400 пациентов с таким диагнозом. Причем каждый пятый из них – в США. Объясняется ли это более отработанной системой диагностики в Соединеных Штатах или распространенностью генетической мутации на том континенте – пока достоверно не ясно.
Это генетическое заболевание, передающееся по аутосомно-рецессивному типу. То есть если оба родителя являются носителями мутации в гене ARSB хромосомы 5, то существует 25-процентная вероятность рождения ребенка с синдромом Марото-Лами. Мутации в гене ARSB приводят к снижению активности лизосомного фермента арилсульфатазы В, отвечающего за расщепление дерматансульфата — важного компонента соединительной ткани. Дерматансульфат содержится в соединительной ткани кожи, сухожилий, кровеносных сосудов, дыхательных путей и клапанов сердца. Если обмен дерматансульфата нарушен, то продукты его распада накапливаются в клетках – и это приводит к развитию патологии.
Синдром МаротоЛами существует в легкой и тяжелой формах. При легкой продолжительность жизни может достигать 50 лет. Летальный исход при тяжелой наступает в подростковом возрасте и связан в большинстве случаев с развитием компрессии спинного мозга за счет сужения позвоночного канала или с сердечно-сосудистыми заболеваниями.
В отличие от большинства мукополисахаридозов, Синдром МаротоЛами не связан с задержкой психического развития. Он проявляется задержкой роста (карликовостью), диспропорциональным строением скелета (короткие туловище и шея, длинные конечности), грубыми чертами лица.
Диагностика
Подозрение на мукополисахаридоз возникает обычно на основании внешних признаков: огрубение черт лица, плоская переносица, задержка роста. Дифференциальный диагноз проводится с системными скелетными дисплазиями и с другими лизосомными болезнями накопления, в первую очередь, с мукополисахаридозами I, II, III и VII типов.
По многим причинам диагностика бывает затруднена.
- Самым показательным при всех мукополисахаридозах считается анализ мочи на экскрецию (выделение) ГАГ (гликозаминогликанов). При синдроме Марото—Лами выделяется дерматансульфат, но не выделяется гепарансульфат (как при МПС I, II, IIIи VII типов). Проблема в том, что дерматансульфат в принципе не выделяется в больших количествах – и зачастую результаты лабораторных тестов выглядят сомнительно, и диагноз своевременно не ставится.
- B лейкоцитах периферической крови определяется низкая активность арилсульфатазы В. Именно этот анализ на сегодняшний день считается «золотым стандартом» при диагностике МПС VI.
- ДНК-диагностика не обязательна для подтверждения диагноза, но может быть рекомендована при неоднозначных результатах биохимических исследований или с целью проведения пренатальной или преимплантационной диагностики в семье.
- В качестве вспомогательного метода для уточнения диагноза назначается ЭКГ. У пациентов с синдромом Марото—Лами часто на кардиограмме отмечаются аномалии, среди которых наиболее значимы синусовая тахикардия, отклонения электрической оси сердца вправо и влево, признаки увеличения предсердий.
Симптомы
Клинически аномалии скелета при синдроме Марото—Лами могут быть заметны с рождения:
- искривление позвоночника и сильные боли в спине,
- дыхание через рот, храп,
- большой язык и очень пухлые губы,
- частые инфекции верхних дыхательных путей,
- хронический насморк
При плановом обследовании ортопеды почти наверняка обнаружат недоразвитие таза и дисплазию головки бедра.
Ближе к году отмечается:
- увеличение печени и селезенки,
- дистрофия зубной эмали, кариес
- тугоподвижностью крупных и мелких суставов.
К двум годам становится все более выражено искривление позвоночника и со всей очевидностью обозначается задержка роста.
Лечение
Для лечения МПС-VI разработана ферменто-заместительная терапия. Наглазим (галсульфаза, galsulfase) — первое и пока единственное лекарственное средство, одобренное для лечения больных с VI типом мукополисахаридоза. Этот препарат (производство компании «Биомарин», США) зарегистрирован и применяется в США с 2005 года, в странах Еросоюза – с 2006, в России – с 2009 (регистрационный номер: ЛСР-005730/09-150709). То есть накоплена более чем 10-летняя практика наблюдений, подтверждающая его эффективность.
Ферменто-заметстительная терапия гальсульфазой не только препятствует накоплению, но и способствует выведению ГАГ из депо, что в конечном итоге приводит к повышению подвижности суставов и – при начале применения в раннем возрасте – замедлят развитие дисплазии скелета.
Многолетние исследования подтвердили, что при начале терапии до 12 лет удается восстановить стабильную работу левого желудочка сердца, однако повреждения сердечных клапанов сохраняются даже при многолетней терапии.
Тем не менее, последние исследования, проводившиеся в том числе и в России, показали, что у некоторый пациентов вырабатываются антитела, нейтрализующие действие гальсульфазы – и таким образом препарат становится неэффективным. То есть ферментозаместительная тепрапия могла бы считаться предпочтительным методом лечения синдрома Марото—Лами, однако она подходит не всем.
Трансплантация стволовых клеток при МПС-VI применяется гораздо реже, чем при МПС-I – из-за значительного дисбаланса рисков и выгоды. Вероятность летального исхода в результате трансплантации колеблется в районе 20%, выраженный эффект подтвержден только в отношении коррекции патологий сердечно-сосудистой системы. И, если при синдроме Гурлер риск оправдан восстановлением когнитивных (интеллектуальных) функций, то у пациентов синдромом Марото—Лами интеллект не нарушен – и, соответственно, весомых аргументов для рискованной операции мало.
Симптоматическая и корригирующая терапия подразумевают применение:
- Гепатопротекторов,
- кардиопрепаратов,
- неспецифических стимуляторов роста,
- средств, направленных на борьбу с остеопорозом
Рекомендуются занятия ЛФК с преимущественным воздействием на опорно-двигательный аппарат (позвоночник и суставы) и общий массаж.
Ребенок с диагнозом МПС-VI должен находиться по диспансерным наблюдением у нескольких специалистов:
- Кардиолога,
- Ортопеда,
- Хирурга,
- Пульмонолога,
- Эндокринолога.
Как с этим жить
Поскольку у детей с синдромом Марото—Лами сохранный интеллект, им, как и здоровым детям, нужны развивающие и обучающие программы. Есть смысл рассмотреть возможности инклюзивного образования.
Поскольку основные проблемы у этих детей связаны с патологией опорно-двигательного аппарата и трудностями передвижения, то для повышения качества их жизни нужно современное обрудование: электрокресла, выпрямители, ходунки. Имеет смысл закрепить поручни вдоль стен дома – чтобы ребенок мог самостоятельно передвигаться в помещении.
Полезные ссылки
- https://health.yandex.ru/pills/naglazim-21939 — наглазим, описание препарата
- https://spiporz.ru/ — Союз пациентов и пациентских организаций по редким заболеваниям.
- https://www.mps-russia.org/ — Межрегиональная Благотворительная Общественная Организация «Общество инвалидов, страдающих синдромом Хантера, другими формами мукополисахаридоза и иными редкими генетическими заболеваниями»
- https://vk.com/club36768044 — Люди маленького роста, объединяемся и общаемся. – Группа ВК
- https://vk.com/club43835220 — Группа для маленьких. – Знакомства в ВК.
Куда обратиться
- Отдел врожденных и наследственных заболеваний у детей с нарушением психики Научно-исследовательского клинического института (НИКИ) педиатрии. — https://www.pedklin.ru/
- Национальный медицинский исследовательский Центр Здоровья Детей — https://nczd.ru/department/nii-pediatrii/
- адаптивно-бытовое оборудование для инвалидов – invaprom.ru
- Городской центр орфанных и других редких заболеваний у детей и подростков (Москва) — https://xn--90adclrioar.xn--p1ai/gorodskoy-tsentr-orfannykh-i-drugikh-redkikh-zabolevaniy-u-detey-i-podrostkov/
- ФГБУ «Научно-исследовательский детский ортопедический институт имени Г.И. Турнера» Министерства здравоохранения РФ (Санкт-Петербург, Пушкин) — https://www.rosturner.ru/
Источник
А
Б
В
Г
Д
Ж
З
И
Й
К
Л
М
Н
О
П
Р
С
Т
У
Ф
Х
Ц
Ч
Ш
Э
Ю
Я
- Что такое Мукополисахаридоз типа VI (синдром Марото-Лами, болезнь Марото-Лами)
- Симптомы Мукополисахаридоза типа VI (синдрома Марото-Лами, болезни Марото-Лами)
- Диагностика Мукополисахаридоза типа VI (синдрома Марото-Лами, болезни Марото-Лами)
- Лечение Мукополисахаридоза типа VI (синдрома Марото-Лами, болезни Марото-Лами)
- Профилактика Мукополисахаридоза типа VI (синдрома Марото-Лами, болезни Марото-Лами)
- К каким докторам следует обращаться если у Вас Мукополисахаридоз типа VI (синдром Марото-Лами, болезнь Марото-Лами)
Что такое Мукополисахаридоз типа VI (синдром Марото-Лами, болезнь Марото-Лами)
Мукополисахаридоз типа VI (синдром Марото — Лами, болезнь Марото — Лами) в 1960 г. впервые описан французскими врачами Марото (Р. Maroteaux) и Лами (М. Е.J. Lamy).
Симптомы Мукополисахаридоза типа VI (синдрома Марото-Лами, болезни Марото-Лами)
Первые симптомы появляются у детей старше 2 лет. Характерно отставание в росте, грубые черты лица (как при синдроме Гурлер, но менее выражены), малые размеры верхней челюсти, короткая шея, бочкообразная грудная клетка, укороченные ключицы. Отмечаются сгибательные контрактуры суставов верхних конечностей (больные не могут поднять руки вверх); с возрастом появляются контрактуры в суставах нижних конечностей, нарушается походка. Часто присоединяются острые респираторные вирусные инфекции. Нередко выявляются грыжи, гепатоспленомегалия. Могут наблюдаться гидроцефалия, спастические параличи. Интеллект не страдает.
По степени выраженности симптомов М. болезнь Марото — Лами делят на типичную классическую форму (А) и легкую, или мягкую (В), с незначительной выраженностью симптомов.
Диагностика Мукополисахаридоза типа VI (синдрома Марото-Лами, болезни Марото-Лами)
При рентгенологическом исследовании выявляются изменения формы позвонков, которые вначале имеют двояковыпуклую форму, а затем приобретают кубовидную или клиновидную; деформации таза: он приобретает форму треугольника, вертлужные впадины мелкие, головки бедренных костей гипоплазированы, треугольной формы или уплощенные. Малые берцовые кости укорочены. Кисти и стопы чаще обычной формы, иногда напоминают изменения при синдроме Гурлер (когтистая лапа) и болезни Моркио (деформация стопы). Отмечается деформация костей верхних и нижних конечностей, ребер, ключиц; возможен асептический некроз головки бедренной кости.
Дифференциальный диагноз проводят с синдромом Гурлер, при котором страдает интеллект.
Лечение Мукополисахаридоза типа VI (синдрома Марото-Лами, болезни Марото-Лами)
Лечение симптоматическое. При этом больных наблюдают разные специалисты — хирурги (удаление грыж), ортопеды (ортопедическая коррекция нарушений опорно-двигательного аппарата), педиатры (в связи с частыми острыми респираторными вирусными инфекциями, сердечно-сосудистой недостаточностью), оториноларингологи (в связи с нарушениями слуха, хроническими отитами и синуситами), офтальмологи, нейрохирурги и невропатологи (внутричерепная гипертензия). Использование для лечения гормональных препаратов (кортикотропина, глюкокортикоидов, тиреоидина), витамина А, переливаний препаратов крови плазмы, введения декстрана-70 приводит лишь к временному улучшению.
Профилактика Мукополисахаридоза типа VI (синдрома Марото-Лами, болезни Марото-Лами)
Профилактика заключается в проведении медико-генетического консультирования и антенатальной диагностики (определение активности ферментов и содержания гликозаминогликанов в культуре клеток амниотической жидкости)
К каким докторам следует обращаться если у Вас Мукополисахаридоз типа VI (синдром Марото-Лами, болезнь Марото-Лами)
Педиатр
Генетик
А
Б
В
Г
Д
Ж
З
И
Й
К
Л
М
Н
О
П
Р
С
Т
У
Ф
Х
Ц
Ч
Ш
Э
Ю
Я
Акции и специальные предложения
Медицинские новости
14.11.2019
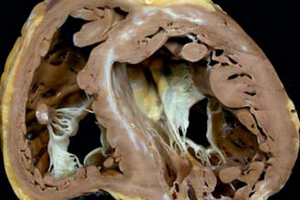
Специалисты сходятся во мнении, что необходимо привлечение внимания общественности к проблемам сердечно-сосудистых заболеваний. Некоторые из них являются редкими, прогрессирующими и трудно диагностируемыми. К таким относится, например, транстиретиновая амилоидная кардиомиопатия
14.10.2019
12, 13 и 14 октября, в России проходит масштабная социальная акция по бесплатной проверке свертываемости крови – «День МНО». Акция приурочена к Всемирному дню борьбы с тромбозами.
Медицинские статьи

Офтальмология является одной из наиболее динамично развивающихся областей медицины. Ежегодно появляются технологии и процедуры, позволяющие получать результат, который еще 5–10 лет назад казался недостижимым. К примеру, в начале XXI века лечение возрастной дальнозоркости было невозможно. Максимум, на что мог рассчитывать пожилой пациент, — это на…
Почти 5% всех злокачественных опухолей составляют саркомы. Они отличаются высокой агрессивностью, быстрым распространением гематогенным путем и склонностью к рецидивам после лечения. Некоторые саркомы развиваются годами, ничем себя не проявляя…
Вирусы не только витают в воздухе, но и могут попадать на поручни, сидения и другие поверхности, при этом сохраняя свою активность. Поэтому в поездках или общественных местах желательно не только исключить общение с окружающими людьми, но и избегать…
Вернуть хорошее зрение и навсегда распрощаться с очками и контактными линзами – мечта многих людей. Сейчас её можно сделать реальностью быстро и безопасно. Новые возможности лазерной коррекции зрения открывает полностью бесконтактная методика Фемто-ЛАСИК.
Источник