Больных с синдромом 47 xyy

 Клиническая генетика. Е.Ф. Давыденкова, И.С. Либерман. Ленинград. «Медицина». 1976 год.
Клиническая генетика. Е.Ф. Давыденкова, И.С. Либерман. Ленинград. «Медицина». 1976 год.
ВЕДУЩИЕ СПЕЦИАЛИСТЫ В ОБЛАСТИ ГЕНЕТИКИ
 Амелина Светлана Сергеевна — профессор кафедры по курсу генетики и лабораторной генетики, доктор медицинских наук. Врач генетик высшей квалификационной категории
Амелина Светлана Сергеевна — профессор кафедры по курсу генетики и лабораторной генетики, доктор медицинских наук. Врач генетик высшей квалификационной категории
Прочитать о докторе подробнее
 Дегтерева Елена Валентиновна — ассистент кафедры по курсу генетики и лабораторной генетики, врач-генетик первой категории
Дегтерева Елена Валентиновна — ассистент кафедры по курсу генетики и лабораторной генетики, врач-генетик первой категории
Прочитать о докторе подробнее
 Редактор страницы: Крючкова Оксана Александровна
Редактор страницы: Крючкова Оксана Александровна
До самого последнего времени выделение этого синдрома представляло большие затруднения. Если остальные клинические синдромы с аномалиями половых хромосом легко выявлялись в общей и отобранной популяциях методом определения полового Х-хроматина, выявление аномалий У-хромосомы было возможно только при кариологическом анализе. В настоящее время в связи с внедрением в клиническую цитогенетику быстрого определения У-хроматина в буквальных мазках методом люминесцентной микроскопии облегчилась диагностика и этих Синдромов.
Частота синдрома 47 у новорожденных мальчиков равна в среднем 1 : 1000 и только по данным американских авторов (Sergovich и др., 1969) необычно высока (1 : 250). Значительное количество больных с этим синдромом было выделено Casey и сотр. (1966) при обследовании специальных больниц особого назначения — 12 мужчин среди 50 с антисоциальным поведением и легкой степенью умственной отсталости и 4 среди 50, которые были госпитализированы по тем же причинам, но имели нормальный интеллект. Для исследования отбирались мужчины высокого роста. По мере накопления наблюдений данные о частоте заболевания будут уточнены.
Клиническая симптоматика этого синдрома, как и синдрома трисомии X, весьма полиморфна, и первоначальные критерии для выделения больных по фенотипу не всегда подтверждаются и, вероятно, во многом будут пересмотрены. Так, высокий рост мужчин с добавочной У- хромосомой, по-видимому, нельзя считать обязательным симптомом. По данным ряда авторов, средний рост больного с кариотипом 47,XYY равен 186 см. Это является важным указанием для отбора на кариотипирование, но не абсолютным, так как в последующих сводках встречаются мужчины с XYY среднего роста.
У описанных больных с добавочной У-хромосомой отмечались психопатические черты характера. Они агрессивны и отличаются неправильным поведением. Наряду с этими характерологическими особенностями
они не обнаруживают значительной задержки умственного развития, а часть больных вообще имеет нормальный интеллект. При обследовании отобранных популяций психиатрических больниц или учреждений для олигофренов редко выявляются больные с синдромом 47ДУУ. Так, большое обследование, проведенное Newton, Jacobs и др. (1972) в психиатрической больнице для умственно отсталых, среди 694 мужчин не выявило ни одного больного с синдромом 47ДУУ, в то время как исследования, проведенные Jacobs с соавт. (1968) в психиатрической больнице, где помещались лица с агрессивным и криминальным поведением, выявили 9 лиц с кариотипом 47ДУУ. Это позволяет считать, что в отличие от синдрома Клайнфельтера добавочная У-хромосома влияет больше на поведение, чем на интеллект.
Соматические аномалии встречаются у небольшой части больных. Встречается евнухоидное строение тела, увеличение нижней челюсти, придающее больным вместе с высоким ростом акромегалоидные черты внешности, неправильный прикус и неправильное расположение зубов, радио-ульнарный синостоз, spina bifida, двусторонняя девиация коленных и локтевых суставов.
У большинства больных с добавочной У-хромосомой не имеется нарушения половой дифференцировки. Эти мужчины фертильны,
Рис. 18. Фенотип (а) и кариотип (б) больного с синдромом XYY.

Синдром 47, XYY

Синдром 47, XYY
и рождающиеся от них дети имеют нормальный кариотип. При обследовании мужнин, страдающих бесплодием, больных с набором половых хромосом типа XYY не было выявлено.
У некоторых больных имеются гормональные изменения: уровень андрогенов значительно выше, чем в норме, так же как уровень лютеинизирующего гормона. Уровень фолликулостимулирующего гормона в пределах нормы,
Таким образом, клиническая симптоматика не дает в настоящее время опорных пунктов для диагноза. Цитогенетическое исследование выявляет наличие двух телец У-хроматина в буквальных мазках и добавочную У-хромосому в кариотипе.
В качестве примера этого синдрома можно привести историю болезни больного, находящегося под наблюдением нашей лаборатории (наблюдение Р. И. Куликова, рис. 18).
Больной Т., 21 год. Плохо учился, отличался агрессивностью и неустойчивым настроением, странностями поведения, по поводу чего был госпитализирован в психиатрическую больницу. Выписался с диагнозом «умеренная степень дебильности». Злоупотребляет алкоголем, привлекался к уголовной ответственности за ряд правонарушений. Больной правильного телосложения. Рост — 187 см, вес — 72 кг. Соматических аномалий и нарушений половой дифференцировки не отмечается.
В.ОК. 25.02.2016г.
ОПТ.ОК. 25.02.2016г.
Источник
XYY-синдром, также известен как YY-синдром или Синдром Джейкобс (d)[1], — хромосомное заболевание, характерное только для мужчин. Носитель синдрома имеет дополнительную Y-хромосому, общий хромосомный набор составляет 44 аутосомы и три половые хромосомы. Внешне мужчины с дополнительной Y-хромосомой обычно не имеют существенных отличий от мужчин с обычным набором хромосом, но могут иметь ряд особенностей[2].
История исследования[править | править код]
Синдром был предсказан в работах Патрисии Джейкобс 1959 года по XXY-синдрому[1], а впервые обнаружен в 1961 году при случайном обследовании мужчины, дети которого имели ряд заболеваний (в частности, один из детей имел синдром Дауна)[3][4]. XYY-синдром был описан как так называемый «синдром сверх-самца» или «синдром сверх-мужчины» (англ. super-male syndrome), при этом носителям синдрома приписывалось агрессивное поведение и тенденция к криминальным действиям. Первые исследователи болезни в 1960-х годах обнаружили относительно высокое количество мужчин с этим синдромом среди обитателей тюрем и психиатрических клиник. Это послужило основой для стереотипа о «сверх-мужчине»[2].
Последующие исследования показали, что абсолютное большинство носителей синдрома никогда не имели отношения к преступности или психическим заболеваниям, но могут иметь повышенный риск проблем с обучением. Дополнительная Y-хромосома сама по себе не ведёт к чрезмерной агрессивности[2].
Генетические особенности[править | править код]
Диаграмма, показывающая формирование синдрома XYY. MI и MII являются стадиями мейоза, тогда как синие и розовые круги являются мужской и женской клетками соответственно, а синие и розовые полоски — Y- и X-хромосомы соответственно. Фиолетовая клетка имеет 2 Y-хромосомы и 1 Х-хромосому из-за слияния с мужской клеткой с 2 Y-хромосомами, что было связано с проблемами деления в MII мужчины
Причиной возникновения синдрома является нерасхождение Y-хромосом в анафазе II[источник не указан 2293 дня] в процессе сперматогенеза. В результате появляется сперматозоид, несущий вторую Y-хромосому, в результате оплодотворения которым появляется ребёнок с 47 хромосомами в кариотипе (47 XYY). Синдром не является наследуемым состоянием, риск рождения второго ребёнка с этим синдромом не выше, чем в среднем в популяции. Частота появления — примерно 1 случай на 1000 мужчин. Большая часть носителей не знает о своей особенности[2].
Несмотря на наличие лишней хромосомы, сперма XYY-мужчины обычно несёт нормальный набор хромосом, ввиду того, что лишняя Y-хромосома элиминируется. Риск появления детей с хромосомными заболеваниями у большинства мужчин с XYY-синдромом не отличается от такого же риска у мужчины с нормальным генотипом. В то же время, число сперматозоидов с хромосомными аномалиями у некоторых мужчин с XYY-синдромом выше, но неизвестно, насколько существенно это повышает риск появления детей с хромосомными болезнями у таких отцов[2].
Фенотипические характеристики[править | править код]
Наличие второй Y-хромосомы в большинстве случаев не ведёт к каким-либо физическим отклонениям. В то же время, многие мужчины с XYY-синдромом имеют одну или несколько особенностей. При рождении они имеют нормальный рост, но часто быстрее растут в детстве. В среднем, во взрослом состоянии носитель выше, чем 75 % мужчин того же возраста. Некоторые мужчины с синдромом XYY имеют небольшие нарушения координации движений, в результате чего могут казаться неуклюжими. Фертильность чаще всего не нарушена, обычно такие мужчины гетеросексуальны и имеют нормальную сексуальную функцию. Тем не менее, описаны случаи существенного снижения фертильности, вплоть до бесплодия. У небольшого числа носителей также повышен уровень половых гормонов, связанных со сперматогенезом, что может вести к бесплодию ввиду нарушения образования спермы. Неизвестно, насколько высоко число случаев бесплодия у мужчин с XYY-синдромом. IQ находится в пределах нормы, но часто несколько ниже, чем у родных братьев и сестёр. Примерно половина носителей имеет проблемы с обучением, в частности, могут быть нарушения речи и чтения. Может быть повышен риск поведенческих проблем, таких как синдром гиперактивности, мужчины с XYY-синдромом часто импульсивны и эмоционально незрелы[2].
См. также[править | править код]
- Синдром 48, XYYY
- Синдром 49, XYYYY
- Синдром Клайнфельтера
- Синдром Шерешевского-Тёрнера
- Трисомия по Х-хромосоме
Примечания[править | править код]
Источник
Что такое XYY-синдром?
XYY–синдром (или также называемый как YY-синдром или Синдром Джейкобса), — это редкое хромосомное заболевание, которое поражает мужчин. Вызван синдром наличием дополнительной Y-хромосомы. Мужчины обычно имеют одну X и одну Y-хромосому. Тем не менее, люди с этим синдромом имеют одну Х и две Y-хромосомы.
По оценкам, болезнь затрагивает примерно 1 из 1000 живорожденных мальчиков.
Пострадавшие люди обычно очень высокие. Многие испытывают серьезные трудности с прыщами в подростковом возрасте. Дополнительные симптомы могут включать неспособность к обучению и поведенческие проблемы, такие как импульсивность. Интеллект обычно находится в нормальном диапазоне, хотя IQ в среднем на 10-15 баллов ниже, чем у братьев и сестер.
В прошлом было много неправильных представлений об этой болезни. Иногда его называли болезнью мужского пола, потому что считалось, что мужчины с этим синдромом чрезмерно агрессивны и им не хватает сочувствия. Недавние исследования показали, что это не так. Хотя люди с синдромом XYY имеют повышенный риск нарушения обучаемости и поведенческих проблем, они не слишком агрессивны и не подвержены повышенному риску какого-либо серьезного психического заболевания.
Поскольку больные мальчики подвержены более высокому риску нарушения обучаемости, им может быть полезна логопедия, репетиторство и общее понимание конкретных проблем, с которыми они сталкиваются. Хотя первые годы в школе могут быть более сложными для мальчиков с XYY-синдромом, они обычно ведут полноценную, здоровую и нормальную жизнь.
Признаки и симптомы XYY-синдрома

Клинические признаки XYY-синдрома часто неуловимы и не обязательно предполагают серьезное хромосомное расстройство. Соответственно, мужчинам с этим условием часто либо не диагностируют синдром, либо диагностируют неправильную патологию.
Наиболее распространенной физическим симптомом является увеличение роста, которое обычно проявляется после 5 или 6 лет и приводит к росту в среднем на около 6 футов и 3 дюйма в зрелом возрасте (см. фото выше).
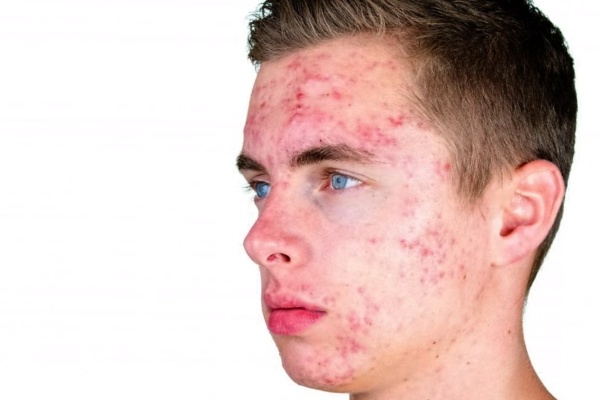
У некоторых людей с XYY-синдромом также развиваются тяжелые кистозные прыщи в подростковом возрасте. Рождаемость и половое развитие нормальны. Помимо возможности увеличения роста, большинство пострадавших людей обычно имеют нормальный внешний вид (фенотип).
Мальчики с синдромом Джейкобса обычно имеют нормальный интеллект, хотя в среднем IQ на 10–15 баллов ниже, чем у его братьев и сестер. У пострадавших мальчиков могут наблюдаться небольшие задержки в достижении основных этапов развития. Проблемы с обучением отмечались в 50% случаев, чаще всего с задержками речи и языковыми проблемами. Проблемы с чтением являются общими из-за повышенной частоты возникновения дислексии.
В некоторых случаях у затронутых людей развиваются поведенческие проблемы, такие как взрывной характер, гиперактивность, импульсивность, вызывающие действия или, в некоторых случаях, антиобщественное поведение.
Существует более высокий уровень дефицита внимания и гиперактивности и меньший повышенный риск развития расстройств аутистического спектра.
Причины XYY-синдрома
XYY-синдром — редкое хромосомное расстройство, вызванное наличием дополнительной Y-хромосомы. Обычно мужчины имеют 46 хромосом, включая одну Х и одну Y-хромосому. Мужчины с XYY-синдромом имеют 47 хромосом, две из которых являются Y-хромосомами.
Большинство случаев XYY-синдрома связаны с ошибкой клеточного деления в сперме до зачатия. Редко ошибка деления клеток возникает после зачатия, что приводит к появлению мозаики клеток с 46 хромосомами и 47 хромосомами.
Точная причина, по которой происходят эти ошибки в делении клеток, не понятна.
Похожие расстройства
Симптомы следующих расстройств могут быть похожи на симптомы XYY-синдрома. Сравнения могут быть полезны для дифференциальной диагностики:
Синдром Клайнфельтера связан с группой хромосомных нарушений у мужчин, в которых присутствует одна или несколько дополнительных Х-хромосом. Мужчины с классической формой расстройства имеют одну дополнительную Х-хромосому. У мужчин с различными формами синдрома Клайнфельтера имеются дополнительные Х и/или Y хромосомы. Дополнительная X и/или Y хромосома может влиять на физическое, развивающее, поведенческое и когнитивное функционирование.
Общие физические особенности могут включать высокий рост, отсутствие вторичного пубертатного развития, небольшие яички (гипогонадизм), замедленное пубертатное развитие и увеличение молочной железы (гинекомастия) в конце полового созревания. Эти особенности могут быть связаны с низким уровнем тестостерона и повышенным уровнем гонадотропина.
Синдром Сотоса (синдром церебрального гигантизма) — переменное генетическое заболевание, характеризующееся чрезмерным ростом до и после рождения. Одной из основных особенностей синдрома Сотоса является особый внешний вид лица, который включает в себя покраснение лица, ненормально выраженный лоб, наклоненные вниз веки, выступающую узкую челюсть, длинное узкое лицо и форму головы похожую на перевернутую грушу.
У большинства детей с синдромом Сотоса присутствуют задержки развития, которые могут включать двигательные и языковые задержки, а также умственную отсталость от легкой до тяжелой степени. Другие проблемы, связанные с синдромом Сотоса, включают желтуху у новорожденных, искривленный позвоночник (сколиоз), эпилепсию, косоглазие, кондуктивную потерю слуха, врожденные пороки сердца, почечные нарушения и поведенческие проблемы. Пострадавшие люди также имеют слегка повышенный риск развития определенных типов опухолей.
Синдром Сотоса вызван аномалией (мутацией) в гене NSD1.
Синдром Марфана — генетическое заболевание, поражающая соединительную ткань, которая является материалом между клетками организма, придающие тканям форму и силу. Соединительная ткань расположена по всему телу, и у людей с синдромом Марфана могут поражаться системы многих органов. Чаще всего поражаются сердечно-сосудистая система, скелетные и глазные системы.
Основные симптомы включают чрезмерный рост костей рук и ног, ненормальное искривление позвоночника из стороны в сторону (сколиоз (см. фото)), вдавливание или выпячивание стенки грудной клетки, вывих линз глаз (смещение хрусталика глаза), близорукость, расширение (аневризма) и разрыв (расслоение) главной артерии, которая носит кровь от сердца (аорты), пролапс митрального клапана и обратный поток крови через аортальный и митральный клапаны (аортальная и митральная регургитация).
Специфические симптомы и степень выраженности синдрома Марфана сильно варьируются от случая к случаю. Синдром Марфана наследуется как аутосомно-доминантный признак.
С синдромом Марфана и связанными с ним расстройствами связаны дефекты или нарушения (мутации) гена фибриллина-1 (FBN1).
Диагностика XYY-синдрома
Диагноз XYY-синдрома ставится на основании тщательной клинической оценки, подробной истории болезни и специальных тестов (т. е. хромосомного анализа), которые выявляют наличие дополнительной Y-хромосомы (47, кариотип XYY).
Диагноз XYY-синдрома может быть поставлен до рождения (внутриутробно) с помощью амниоцентеза или взятия проб ворсин хориона. Во время амниоцентеза образец жидкости, который окружает развивающийся плод, удаляется и анализируется, в то время как взятия проб ворсин хориона включает в себя удаление образцов ткани из части плаценты. Хромосомные исследования, выполненные на таких образцах жидкости или тканях, могут выявить присутствие дополнительной Y-хромосомы.
— Клиническое тестирование и обследование
Оценка речи и языка должна проводиться в течение первых 24 месяцев. Оценка чтения должна проводиться в школьном возрасте, чтобы исключить дислексию. Поведенческая оценка должна рассматриваться для детей, которые испытывают трудности с такими симптомами, как импульсивность и плохое внимание.
Лечение XYY-синдрома
Лечение XYY-синдрома является симптоматическим и поддерживающим. Могут быть полезны логопедия, трудотерапия или помощь в обучении.
В большинстве случаев пострадавшие люди очень чутко реагируют на раннее вмешательство и лечение, и проблемы могут полностью решится в течение нескольких лет.
Лечение прыщей может помочь пострадавшему с самооценкой.
Дефицит внимания и гиперактивность, трудности с социальным взаимодействием или другие поведенческие проблемы можно лечить с помощью терапии или медикаментов так же, как у людей, у которых нет XYY.
Источник
Хромосомные болезни этогруппа патологических состояний, обусловленных мутационными изменениями в хромосомном наборе (таблица 4).
Таблица 4
Частота встречаемости заболеваний, вызванных различными типами
анеуплоидии у человека
| Тип мутации | Синдромы | Частота среди новорожденных |
| Аутосомы | ||
| Трисомия 21 47,XX(XY)+21 | Дауна | 1/700 |
| Трисомия 13 47, XX(XY)+13 | Патау | 1/5 000 |
| Трисомия 18 47, XX(XY)+18 | Эдвардса | 1/10 000 |
| Половые хромосомы (женские) | ||
| ХО, Моносомия 45, XО | Шерешевского-Тернера | 1/500 |
| ХХХ, Трисомия 47, XXX | ХХХ-синдром | 1/700 |
| Половые хромосомы (мужские) | ||
| ХХУ 47, XXY | Клайнфельтера | 1/500 |
| ХХУУ 48, XXY | Клайнфельтера | 1/500 |
| ХУУ 47, XYY | Дубль У | 1/1 000 |
Показано, что примерно у 40% спонтанных абортов и 6% всех мертворожденных имеются хромосомные изменения. В то же время, около 6 из 1000 новорожденных имеют хромосомные нарушения, а удельный вес хромосомных болезней в группе детей с врожденными аномалиями составляет около 50%. Клинически почти все хромосомные болезни проявляются нарушением интеллектуального развития; множественными врожденными пороками. Это может быть умственное и физическое недоразвитие, пороки развития скелета, деформация черепа, микроцефалия, эпикант и мн. др.
Механизм возникновения геномных мутаций связан с патологией нарушения нормального расхождения хромосом в мейозе (анафаза-I и анафаза-II), в результате чего образуются аномальные гаметы (по количеству хромосом), после оплодотворения которых возникают гетероплоидные зиготы (рис. 18).
| Рис. 18. Схематическое изображение нерасхождения одной пары хромосом в I мейотическом делении (Н.П. Бочков и др., 1984); А – мейотическое деление I и II; Б – зиготы: 1 – трисомия, 2 – моносомия |
Хромосомные мутации (хромосомные перестройки, хромосомные аберрации) приводят к изменению числа, размеров и организации хромосом. В случае гетероплоидии особенно тяжелы моносомии. Моносомии по аутосомам заканчиваются летально еще в первые дни эмбрионального развития или приводят к гибели зародыша на более поздних стадиях (спонтанные аборты). Полные трисомии описаны у человека по большому количеству хромосом: 8, 9, 13, 14, 18, 21, X, Y. Наиболее изученными синдромами, в основе которых лежат нарушения в системе аутосом (геномные мутации, хромосомные мутации) являются трисомии 21, 13, 18, транслокационная форма Дауна, синдром «кошачьего крика», в системе половых хромосом трисомии XXY, XXX, XYY и моносомия XO.
Болезнь Дауна (трисомия 21; 47,XX(XY)+21)
Диагностика болезни Дауна уже у новорожденного не вызывает затруднений. При болезни Дауна встречается от 9 до 29 соматических аномалий. Наиболее часто при этом синдроме встречаются: брахицефальный череп со сглаженным затылком и уплощенным лицом, эпикант; пятна Брушфильда (светлые пятна на радужке); маленькие недоразвитые ушные раковины; увеличенный «складчатый» язык; широкие кисти с короткими пальцами и укороченными искривленными пятыми пальцами (клинодактилия); поперечная борозда на одной или обеих ладонях («обезьянья складка»); расширенные промежутки между 1 и 2-м пальцами стоп. Интеллектуальный дефект больных углубляется с возрастом. Известно, что примерно у 60% детей с болезнью Дауна имеются разные формы глазной патологии а у 70% обнаруживают тугоухость.
Большое внимание в последние годы уделяется изучению патогенеза синдрома Дауна. В настоящее время предложена объединенная генетическая гипотеза синдрома Дауна и болезни Альцгеймера. В статусе таких больных выявляется преждевременное старение, преобладание дегенеративных сосудистых нарушений, сахарный диабет, катаракта, липофусциноз, амилоидоз, избирательное повреждение холинергических нейронов в базальных ганглиях, склонность к злокачественным новообразованиям, специфические нарушения слуха и другие признаки, а главное – характерные нарушения интеллекта, напоминающие таковые при старческой болезни Альцгеймера.
Использование цитогенетических методов исследования показало, что примерно 80% всех случаев простой трисомии 21 имеет материнское происхождение и около 20% – отцовское. При этом лишь 20% всех случаев «материнского» синдрома Дауна обусловлено нерасхождением хромосом 21-ой пары во втором делении мейоза, а остальные – ошибками первого деления мейоза.
Болезнь Дауна транслокационной формы (46,XX(XY)t14(13,15,22)/21)
Транслокационные формы синдрома Дауна наблюдаются в 3-4% случаев. Число хромосом в данном варианте болезни нормальное – 46, так как дополнительная хромосома 21 транслоцирована на одну из аутосом (13, 14, 15 или 22) (рис.19).
| Рис. 19 Транслокация 14/21. |
При этом один из фенотипически здоровых родителей является носителем сбалансированной транслокации. В кариотипе этих родителей насчитывается 45 хромосом, а одна из аутосом состоит как бы из двух частей и содержит генетический материал недостающей хромосомы, поэтому при общем числе хромосом, равном 45, нет утери генетического материала (рис. 20). Примерно в 1/3 всех случаев транслокационный вариант синдрома Дауна имеет наследственный характер. Выявление у кого-либо из родителей сбалансированной транслокации определяет необходимость пренатальной диагностики.
Синдром Эдвардса (трисомия 18; 47, XX(XY)+18 )
При кариологическом обследовании больных выявляется лишняя хромосома из группы Е (хромосома 18). Фенотипические проявления синдрома Эдвардса довольно характерны. Это наличие долихоцефального черепа, сдавленного с боков, с низким лбом и широким выступающим затылком; глазные щели узкие; эпикант; нижняя челюсть маленькая, скошена назад (микроретрогнатия); рот маленький, треугольной формы с короткой верхней губой; шея короткая, с крыловидной складкой.
При данном синдроме типичны аномалии опорно-двигательного аппарата: кисти и пальцы короткие, пятые пальцы искривлены, пальцы сжаты в кулак, второй и пятый пальцы расположены сверху и прикрывают прижатые к ладони второй и четвертый пальцы; первый палец стопы короткий и широкий, синдактилия второго и третьего пальцев; форма стопы в виде «качалки».
Почти 95% больных имеют пороки сердца, крупных сосудов, мочеполовой системы, аномалии органов пищеварения. Прогноз для жизни неблагоприятный.
Рис.20 Схема болезни Дауна транслокационной формы
Синдром Патау (трисомия 13; 47, XX(XY)+13 )
При кариологическом анализе соматических клеток больных выявляется лишняя хромосома из группы D (хромосома 13). Клиническая картина типична: микроцефальный череп с низким скошенным лбом и вдавленными височными областями; глазные щели узкие, расположены горизонтально, расстояние между ними уменьшено (гипотелоризм), почти всегда встречается глазная патология; ушные раковины расположены низко, маленькие мочки прижаты к голове, завитки неправильной формы; череп с углублениями в теменно-затылочной области, расстояние между теменными буграми увеличено.
Демонстративным признаком синдрома Патау являются «заячья губа» и «волчья пасть». Расщелины могут быть как двусторонними, так и односторонними. Почти всегда расщепление верхней губы сопровождается расщелиной неба.
Характерны также такие аномалии костно-мышечной системы, как полидактилия на верхних и нижних конечностях, второй и четвертый пальцы согнуты, приведены к ладони и перекрыты первым и пятым пальцами. Выявляются дефекты развития практически всех систем и органов. Мозг часто не разделен на полушария, наблюдается гипоплазия лобных долей, мозжечка.
У 50% больных выявляются пороки развития мочевыводящих путей: кистозная почка, гидронефроз, дисплазия почек, у 50% девочек находят удвоение влагалища и двурогую матку с гипоплазией яичников. Прогноз для жизни неблагоприятный.
Синдром «кошачьего крика» (синдром 5р–, 46XX(XY)del(5р–))
Наиболее частый из всех синдромов делеции аутосом – синдром делеции короткого плеча хромосомы 5. У больных при кариологическом анализе обнаруживается укорочение короткого плеча одной из хромосом группы В.
Фенотипическими признаками синдрома являются: микроцефалия; круглое «лунообразное» лицо в первые годы жизни и узкое лицо в более старшем возрасте; антимонголоидный разрез глаз, эпикант, косоглазие, катаракта, очаги пигментации сетчатки, атрофия зрительных нервов; плоская спинка носа, высокое небо; ушные раковины деформированы; синдактилия пальцев ног, косолапость, мышечная гипотония. Своеобразный симптом – плач при рождении, напоминающий крик кошки. Он присутствует у детей первого года жизни и обусловлен нарушением деятельности центральной нервной системы и изменениями гортани (уменьшение надгортанника, сужение гортани, отечность слизистой оболочки).
Прогноз для жизни зависит от выраженности симптомов. Многие больные доживают до подросткового возраста. Умственная отсталость всегда глубокая. Окончательный диагноз устанавливается в результате исследования кариотипа.
Синдром Шерешевского-Тернера (моносомия Х; 45, XО )
Для синдрома характерно отсутствие в кариотипе половой Х-хромосомы. Частота встречаемости 1:3000, среди девочек, страдающих олигофренией – 1:1500. Частота синдрома возрастает среди низкорослых женщин с недоразвитием вторичных половых признаков и аменореей.
Большинство больных с синдромом Шерешевского–Тернера имеют нормальный или близкий к норме интеллект, но умственная отсталость у них встречается чаще, чем в общей популяции. Интеллектуальные нарушения обычно сочетаются с недоразвитием эмоционально-волевой сферы: больные повышенно внушаемы, несколько некритичны, упрямы, часто эйфоричны.
Диагностика синдрома возможна уже в период новорожденности, так у новорожденной отмечается отечность кистей и стоп, низкий рост волос на шее, шея короткая с крыловидными складками, идущими от сосцевидных отростков к плечам. Характерна чрезмерная подвижность кожи на шее. Отмечаются множественные аномалии развития: эпикант, антимонголоидный разрез глаз; низко расположенные ушные раковины; гипомимия («лицо сфинкса»); высокое небо, аномалии зубов.
Характерны разнообразные скелетные нарушения, например «щитообразная» широкая грудная клетка, гипоплазия или сращение I и II шейных позвонков, широкие кисти с короткими IV и V пальцами, деформация локтевых и коленных суставов, укороченные III и IV пальцы стоп, синдактилия.
Важными диагностическими признаками являются также врожденные пороки сердца, низкий рост (в 98% случаев), половой инфантилизм с первичной аминореей, часты гипоплазия или гипертрофия ногтевых пластинок, гиперпигментация кожи. Наблюдаются дефекты зрения (22%) и слуха (52%).
Офтальмологическое обследование выявляет бледность сосков зрительного нерва, микрофтальм, катаракту, сужение артерий глазного дна. Дерматоглифическое исследование выявляет изменение кожных узоров пальцев и ладоней.
Диагноз может быть установлен с помощью цитологического метода исследования полового хроматина и кариологического анализа.
Синдром трисомии (47, XXX )
Для синдрома характерно наличие в кариотипе дополнительных Х-хромосом. Частота трисомии Х среди новорожденных девочек 1:800. Частота возрастает среди пациенток психиатрических больниц. В период новорожденности и детства редко можно выявить какие-либо фенотипические особенности, имеющие диагностическое значение. Основная психопатологическая особенность синдрома – проявление эмоциональной незрелости и эмоционально-поведенческие нарушения с невротическими и неврозоподобными расстройствами, иногда со склонностью к аутоагрессии. В раннем возрасте характерно выраженное отставание в развитии речи. У женщин с трисомией Х часто наблюдается эндокринный дисбаланс, бесплодие, преждевременный климакс. Могут наблюдаться более сложные полисомии Х: тетрасомия (ХХХХ) и пентасомия (ХХХХХ). Считается, что степень психического недоразвития коррелирует с числом дополнительных Х-хромосом. У женщин с полисомией Х увеличена частота психических заболеваний (шизофрения, эпилепсия, маниакально-депрессивный психоз). Окончательный диагноз устанавливается на основании цитологического обследования щечного эпителия в результате обнаружения полового хроматина в кариотипе.
Синдром Клайнфельтера (47, XXY )
Для синдрома характерно наличие в кариотипе мужчины дополнительной половой Х-хромосомы. Частота синдрома составляет в среднем 1 на 850 новорожденных мужского пола и 1–2.5% у больных олигофренией в степени дебильности. Клинические проявления достаточно вариабельны. Обязательными диагностическими критериями являются гипогенитализм и гипогонадизм. Характерными признаками также являются: высокий рост, высокое стояние таза, евнухоидные пропорции, астеническое телосложение, узкие плечи, удлиненные конечности.
Мышечная система развита слабо. Это особенно четко проявляется в препубертантном и пубертантном возрасте. У взрослых нередко встречаются склонность к ожирению по женскому типу, гинекомастия, слабое подмышечное оволосение, оволосение на лобке по женскому типу. Отмечают недоразвитие вторичных половых признаков с гипоплазией яичек и часто полового члена. Мужчины с синдромом Клайнфельтера бесплодны. Частыми являются различные диспластические признаки: брахицефалия; низкий рост волос на затылке, уплощенный затылок; гипертелоризм; эпикант; деформация ушных раковин; выступающие надбровные дуги; аномалии зубов; искривление и укорочение V пальцев.
Болезнь часто сопровождается задержкой психического развития. Диагноз может быть установлен на основании кариологического анализа, обнаружения полового хроматина в щечном эпителии.
Синдром дубль Y (47, XYY )
Синдром характеризуется наличием в кариотипе дополнительной Y-хромосомы. Наблюдается у мальчиков и мужчин высокого роста. Частота среди новорожденных мальчиков 1:840. Выраженных нарушений фенотипа может не наблюдаться. Примерно у 80% лиц с данным синдромом наблюдаются признаки психического недоразвития в сочетании с нарушениями эмоционально-волевой сферы и поведения. Больные испытывают трудности в социальной адаптации. Многим характерны замедленность и ригидность мышления, речи и моторики, часто снижена способность к самокритике. Наблюдается сочетание умственной отсталости с психопатоподобным поведением, агрессивностью, расторможенностью и извращением влечения. Отмечаются самоуверенность, импульсивность, гиперсексуальность. Окончательный диагноз устанавливается при цитологическом обследовании.
Высказывается предположение, что психопатоподобные формы поведения при наличии несбалансированного кариотипа по половым хромосомам связаны с вторичными изменениями в деятельности нервной системы как следствие нарушений гормональной сферы.
Источник