Болезнь шарко мари код мкб
Утратил силу — Архив
![]()
РЦРЗ (Республиканский центр развития здравоохранения МЗ РК)
Версия: Архив — Клинические протоколы МЗ РК — 2010 (Приказ №239)
Категории МКБ:
Наследственная моторная и сенсорная невропатия (G60.0)
Общая информация
Краткое описание
Наследственные невропатии представляют собой гетерогенную группу генетически детерминированных заболеваний нервной системы, проявляющихся множественным поражением периферических нервов и различающихся типом наследования, выраженностью клинического полиморфизма, особенностями течения, характером электромионейрографических и морфогистохимических изменений.
Протокол «Наследственная моторная и сенсорная невропатия»
Коды по МКБ-10: G 60,0
— Болезнь Шарко-Мари Тута (наследственная мотосенсорная невропатия, тип I)
— Болезнь Дежерина — Сотта (наследственная мотосенсорная невропатия, тип III)
— Наследственная моторная и сенсорная невропатия, типы I-IV
— Гипертрофическая невропатия у детей
— Перонеальная мышечная атрофия (аксональный тип, гипертрофический тип)
— Синдром Руси-Леви
Мобильное приложение «MedElement»
— Профессиональные медицинские справочники. Стандарты лечения
— Коммуникация с пациентами: вопросы, отзывы, запись на прием
Скачать приложение для ANDROID / для iOS
Мобильное приложение «MedElement»
— Профессиональные медицинские справочники
— Коммуникация с пациентами: вопросы, отзывы, запись на прием
Скачать приложение для ANDROID / для iOS
Классификация
Клинико-генетическая классификация наследственных мотосенсорных невропатий
Аутосомно-доминантная наследственная мотосенсорная невропатия, тип I:
Тип I А:
— обусловленная дупликацией 17 р 11.2;
— обусловленная точковыми мутациями гена миелина РМР-22.
Тип I В: обусловленная точковыми мутациями гена миелина Ро.
Тип I С: с неидентифицированным генетическим дефектом.
Аутосомно-рецессивная наследственная мотосенсорная невропатия, тип I: с неидентифицированным генетическим дефектом.
Аутосомно-доминантная наследственная мотосенсорная невропатия, тип II: с неидентифицированным генетическим дефектом.
Аутосомно-рецессивная наследственная мотосенсорная невропатия, тип II: с неидентифицированным генетическим дефектом.
Наследственная мотосенсорная невропатия, тип III:
— обусловленная дупликацией 17 р 11.2;
— обусловленная точковыми мутациями гена миелина РМР-22;
— обусловленная точковыми мутациями гена миелина Ро.
Х- сцепленная наследственная мотосенсорная невропатия:
— обусловленная точковыми мутациями и делециями коннексина 32;
— с неидентифицированным генетическим дефектом.
Сложные формы наследственных мотосенсорных невропатий:
— с пирамидными симптомами;
— с атрофией зрительных нервов;
— с атрофией зрительных нервов и глухотой;
— с пигментной ретинопатией;
— с кератодермией ладоней и стоп.
Диагностика
Диагностические критерии
Жалобы и анамнез: мышечная утомляемость, атрофии, боли в дистальных отделах конечностей по «полиневритическому типу», костные деформации, нарушение походки, задержка моторного развития, прогрессирующее течение.
В анамнезе: при болезни Шарко-Мари — аутосомно-рецессивный тип наследования, дебют заболевания в первое десятилетие жизни.
При болезни Дежерина-Сотта (наследственной мотосенсорной невропатии, тип III): аутосомно-рецессивный тип наследования, дебют в первые два года жизни, задержка моторного развития, слабость и атрофии дистальных отделов конечностей, по мере прогрессирования — поражение проксимальных отделов, атаксия, быстропрогрессирующее течение.
Для наследственной мотосенсорной невропатии, тип I характерен аутосомно-доминантный тип наследования, дебют заболевания преимущественно в 1-2 десятилетие жизни, медленно-прогрессирующий характер течения.
Для наследственной мотосенсорной невропатии, тип II — характерен аутосомно-доминантный тип наследования, дебют во второй декаде жизни, отсутствие либо незначительно выраженные атрофии дистальных отделов конечностей, относительно доброкачественное течение.
Для наследственной мотосенсорной невропатии, тип II — характерен аутосомно-рецессивный тип наследования, дебют заболевания в раннем возрасте, выраженные атрофии и слабость дистальных отделов конечностей, деформации кистей и стоп, быстропрогрессирующий характер течения.
Физикальное обследование: неврологический статус — боли (ноющие, стреляющие, крампи) в дистальных отделах конечностей на фоне гипостезии, с постепенным присоединением слабости, атрофии в дистальных отделах, снижение и утрата ахилловых рефлексов по мере прогрессирования — карпо-радиальных; нарушение походки по типу «петушиной», степпаж.
Атрофии возникают в дистальных отделах с последующей (при прогрессировании) атрофией проксимальных отделов конечностей. Деформированные стопы по типу «клюшки» или «фридрейховской» стопы в виде полой стопы с высоким сводом, экстензией основных фаланг. При дебюте заболевания в детском возрасте — задержка моторного развития.
Лабораторные исследования: общий анализ крови и мочи без патологии.
Инструментальные исследования
ЭМГ различна: для наследственной мотосенсорной невропатии, тип I характерно снижение скорости проведения импульса по моторным и сенсорным волокнам.
Для болезни Шарко-Мари снижение скорости проведения импульса по моторным и сенсорным волокнам.
Для болезни Дежерина-Сотта значительное снижение (менее 12 м/с) скорости проведения импульса по периферическим нервам.
Для наследственной мотосенсорной невропатии, тип II (аутосомно-рецессивная форма) на ЭМГ характерно значительное снижение скорости проведения импульса по периферическим нервам (менее 38 м/с).
Для наследственной мотосенсорной невропатии, тип II (аутосомно-доминантная форма) умеренное снижение скорости проведения импульса по периферическим нервам.
Показания для консультации специалистов:
1. Психолог.
2. Логопед.
3. Ортопед.
4. Генетик.
5. Протезист.
Минимум обследования при направлении в стационар:
1. Общий анализ крови и мочи.
2. АЛТ.
3. АСТ.
4. Кал на яйца глист.
Основные диагностические мероприятия:
1. Общий анализ крови.
2. Общий анализ мочи.
3. ЭМГ.
4. Логопед.
5. Психолог.
6. Окулист.
7. Протезист.
8. Ортопед.
Дополнительные диагностические мероприятия:
1. ЭКГ.
2. Кардиолог.
Дифференциальный диагноз
Дифференциально-диагностические признаки аутосомно-доминантных наследственных мотосенсорных невропатий
Признак | Аутосомно-доминантная наследственная мотосенсорная невропатия | |
Тип I | Тип II | |
Дебют заболевания | Чаще первая декада жизни (60% больных) | Чаще вторая декада жизни (70% больных) |
Мышечная слабость | Прогрессирует с возрастом | Может быть не выражена |
Деформация стоп | Наблюдается в 2/3 случаях | Наблюдается в 1/3 случаях |
Сухожильные рефлексы | Резкое снижение пяточных рефлексов | Возможно снижение пяточных рефлексов |
Чувствительные расстройства | Постоянны | Возможны |
Тремор кистей | Часто | Редко |
Скорость проведения импульса по срединному нерву | Менее 30 м/с | Более 40 м/с |
Дифференциально-диагностические признаки наследственных нервно-мышечных заболеваний
Признак | Невральная амиотрофия | Спинальная амиотрофия | Миопатии |
Дебют заболевания | Первая декада жизни | Первая декада жизни | Первая декада жизни |
Мышечная слабость | Прогрессирует с возрастом | Прогрессирует с возрастом | Прогрессирует с возрастом |
Чувствительные расстройства | Постоянны | Не характерны | Не характерны |
Деформация стоп | Наблюдается в 2/3 случаях | Не характерны | Не характерны |
Сухожильные расстройства | Резкое снижение пяточных рефлексов | Гипо-, арефлексия | Гипо-, арефлексия |
Патология сердца | Не характерна | Не характерна | Миокардиодистрофия |
ЭМГ | I тип, характерно снижение скорости проведения импульса по моторным и сенсорным волокнам | II тип, денервационный характер изменений | Первично-мышечный характер изменений |
Лечение
Тактика лечения
Цели консервативного лечения:
1. Коррекция двигательных нарушений.
2. Коррекция нервно-психического статуса пациентов.
3. Обеспечить больному социальную адаптацию.
4. Определить форму невральной амиотрофии и обеспечить адекватное лечение.
Немедикаментозное лечение:
1. Физиотерапия.
2. Лечебная физкультура.
3. Кондуктивная педагогика.
4. Массаж.
5. Ортопедическая коррекция.
Медикаментозное лечение:
1. Антиоксидатная терапия:
— никотинамид 10-20 мг/сут.;
— аевит 1 кап./сут.
2. Общеукрепляющая терапия: витамины группы В, фолиевая кислота, препараты магния.
3. Аминокислоты: церебролизин, метионин, глутаминовая кислота.
4. Препараты, влияющие на тканевой метаболизм: рибоксин, карнитин, коэнзим Q, убихинон.
5. Препараты, улучшающие периферическое кровообращение: трентал, теоникол.
6. Антихолинэстеразные препараты: прозерин, нейромидин.
Профилактические мероприятия: полноценное белковое питание с ограничением углеводов, жиров. Санация очагов хронической инфекции. Профилактика вирусных и бактериальных инфекций.
Дальнейшее ведение: диспансерное наблюдение невропатолога по месту жительства, оформление пособия по инвалидности.
Основные медикаменты:
1. L- карнитин, таблетки
2. Аевит, капсулы
3. Глутаминовая кислота, таблетки 0,5
4. Карнитина хлорид, флакон 20 % — 100
5. Магне В6, таблетки
6. Никотинамид, ампула 1% -1,0; 2,5% — 1,0
7. Никотинамид, таблетки 0,005
8. Пентоксифиллин (трентал), таблетки 0,1
9. Пиридоксин гидрохлорид, ампулы, 1 мл 5%
10. Прозерин, ампулы 0,05% 1 мл
11. Тиамин хлорид, ампулы 5% 1 мл
12. Фолиевая кислота, таблетки 0,001
13. Церебролизин, ампулы 1 мл
14. Цианокобаламин, ампулы 1 мл 200 мкг и 500 мкг
Дополнительные медикаменты:
1. Дибазол, таблетки 0,02, 0,005
2. Коэнзим Q
3. Ксантинола никотинат (теоникол), таблетки 0,15
4. Метионин, таблетки 0,25
5. Нейромидин, таблетки 20 мг
6. Нейромультивит, таблетки
7. Рибоксин, таблетки 0,2
Индикаторы эффективности лечения:
— улучшение самочувствия;
— уменьшение мышечной слабости;
— повышение эмоционального и психического тонуса;
— увеличение объема активных движений;
— уменьшение выраженности чувствительных расстройств.
Госпитализация
Показания к плановой госпитализации: прогрессирующая мышечная слабость, атрофии мышц, деформации конечностей, ограничение активных движений, мышечная гипотония, нарушение походки, расстройства чувствительности, боли, парестезии в конечностях.
Информация
Источники и литература
- Протоколы диагностики и лечения заболеваний МЗ РК (Приказ №239 от 07.04.2010)
- Петрухин А.С. Неврология детского возраста, Москва 2004
Наследственные болезни нервной системы. Под редакцией Ю.Е. Вельтищева, П.А. Темина, Москва 1998 г.
Неврология. Под редакцией М. Самуэльса, Москва 1997 г.
- Петрухин А.С. Неврология детского возраста, Москва 2004
Информация
Список разработчиков:
№ | Разработчик | Место работы | Должность |
1. | Мухамбетова Гульнара Амерзаевна | КазНМУ, кафедра нервных болезней | Ассистент, кандидат медицинских наук |
2. | Кадыржанова Галия Баекеновна | РДКБ «Аксай», психоневрологическое отделение №3 | Заведующая отделением |
3. | Серова Татьяна Константиновна | РДКБ «Аксай», психоневрологическое отделение №1 | Заведующая отделением |
4. | Балбаева Аиым Сергазиевна | РДКБ «Аксай», психоневрологическое отделение №3 | Врач-ординатор |
Прикреплённые файлы
Внимание!
Если вы не являетесь медицинским специалистом:
- Занимаясь самолечением, вы можете нанести непоправимый вред своему здоровью.
- Информация, размещенная на сайте MedElement и в мобильных приложениях «MedElement (МедЭлемент)», «Lekar Pro»,
«Dariger Pro», «Заболевания: справочник терапевта», не может и не должна заменять очную консультацию врача.
Обязательно
обращайтесь в медицинские учреждения при наличии каких-либо заболеваний или беспокоящих вас симптомов.
- Выбор лекарственных средств и их дозировки, должен быть оговорен со специалистом. Только врач может
назначить
нужное лекарство и его дозировку с учетом заболевания и состояния организма больного.
- Сайт MedElement и мобильные приложения «MedElement (МедЭлемент)», «Lekar Pro»,
«Dariger Pro», «Заболевания: справочник терапевта» являются исключительно информационно-справочными ресурсами.
Информация, размещенная на данном
сайте, не должна использоваться для самовольного изменения предписаний врача.
- Редакция MedElement не несет ответственности за какой-либо ущерб здоровью или материальный ущерб, возникший
в
результате использования данного сайта.
Источник
Связанные заболевания и их лечение
Описания заболеваний
Стандарты мед. помощи
Содержание
- Описание
- Дополнительные факты
- Классификация
- Симптомы
- Диагностика
- Лечение
Названия
Название: Невральная амиотрофия Шарко-Мари-Тута.
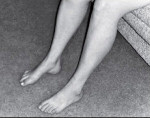
Невральная амиотрофия Шарко-Мари-Тута
Описание
Невральная амиотрофия Шарко. Мари — Тута — прогрессирующее хроническое наследственное заболевание с поражением периферической нервной системы, приводящем к мышечным атрофиям дистальных отделов ног, а затем и рук. Наряду с атрофиями наблюдается гипестезия и угасание сухожильных рефлексов, фасцикулярные подергивания мышц. К диагностическим мероприятиям относятся электромиография, электронейрография, генетическое консультирование и ДНК-диагностика, биопсия нервов и мышц. Лечение симптоматическое — курсы витаминотерапии, антихолинэстеразной, метаболической, антиоксидантной и микроциркуляторной терапии, ЛФК, массажа, физиопроцедур и водолечение.
Дополнительные факты
Невральная амиотрофия Шарко-Мари-Тута (ШМТ) относится к группе прогрессирующих хронических наследственных полиневропатий, в которую входят синдром Русси-Леви, гипертрофическая невропатия Дежерина-Сотта, болезнь Рефсума и другие более редкие заболевания. Болезнь Шарко-Мари-Тута характеризуется аутосомно-доминантным наследованием с пенетрантностью на уровне 83%. Встречаются также случаи аутосомно-рецессивного наследования. Лица мужского пола болеют чаще, чем женщины.
По различным данным невральная амиотрофия Шарко-Мари-Тута встречается с частотой от 2 до 36 случаев на 100 тыс. Населения. Зачастую болезнь носит семейный характер, причем у членов одной семьи клинические проявления могут иметь различную выраженность. Наряду с этим наблюдаются и спорадические варианты ШМТ.
Отмечена связь болезни Шарко-Мари-Тута и атаксии Фридрейха. В отдельных случаях у пациентов с ШМТ со временем отмечаются типичные признаки болезни Фридрейха и наоборот — иногда по прошествии многих лет клиника атаксии Фридрейха сменяется симптоматикой невральной амиотрофии. Некоторыми авторами даны описания промежуточных форм этих заболеваний. Наблюдались случаи, когда у одних членов семьи диагностировалась атаксия Фридрейха, а у других — амиотрофия ШМТ.
Патогенетические аспекты.
На сегодняшний день неврология как наука не располагает достоверными сведениями о этиологии и патогенезе невральной амиотрофии. Проведенные исследования показали, что у 70-80% пациентов с ШМТ, прошедших генетическое обследование, отмечалось дублирование определенного участка 17-й хромосомы. Определено, что невральная амиотрофия Шарко-Мари-Тута имеет несколько форм, вероятно обусловленных мутациями различных генов. Например, исследователи выяснили, что при форме ШМТ, вызванной мутацией кодирующего митохондриальный белок гена MFN2, происходит образование сгустка митохондрий, нарушающего их продвижение по аксону.
Установлено, что большинство форм ШМТ связаны с поражением миелиновой оболочки волокон периферических нервов, реже встречаются формы с патологией аксонов — осевых цилиндров проходящих в центре нервного волокна. Дегенеративные изменения затрагивают также передние и задние корешки спинного мозга, нейроны передних рогов, пути Голля (спинномозговые проводящие пути глубокой чувствительности) и столбы Кларка, относящиеся к заднему спинномозжечковому пути.
Вторично, в результате нарушения функции периферических нервов, развиваются мышечные атрофии, затрагивающие отдельные группы миофибрилл. Дальнейшее прогрессирование болезни характеризуется смещением ядер сарколеммы, гиалинизацией пораженных миофибрилл и интерстициальным разрастанием соединительной ткани. В последующем нарастающая гиалиновая дегенерация миофибрилл приводит к их распаду.
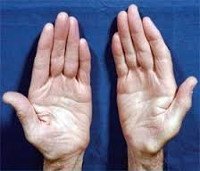
Невральная амиотрофия Шарко-Мари-Тута
Классификация
В современной неврологической практике невральная амиотрофия Шарко-Мари-Тута подразделяется на 2 типа. Клинически они являются практически однородными, однако имеют ряд особенностей, позволяющих провести такое разграничение. Невральная амиотрофия I типа характеризуется существенным снижением скорости проведения нервного импульса, при ШМТ II типа скорость проведения страдает незначительно. Биопсия нерва обнаруживает при I типе сегментарную демиелинизацию нервных волокон, гипертрофический рост непораженных шванновских клеток; при II типе — дегенерацию аксонов.
Симптомы
Невральная амиотрофия Шарко-Мари-Тута начинается с развития симметричных мышечных атрофий в дистальных отделах ног. Начальные симптомы манифестируют, как правило, в первой половине второго десятилетия жизни, реже в период от 16 до 30 лет. Они заключаются в повышенной утомляемости стоп при необходимости длительно стоять на одном месте. При этом наблюдается симптом «топтания» — чтобы снять утомляемость стоп пациент прибегает к ходьбе на месте. В отдельных случаях невральная амиотрофия манифестирует расстройствами чувствительности в стопах, наиболее часто — парестезиями в виде ползания мурашек. Типичным ранним признаком ШМТ является отсутствие ахилловых, а позже и коленных сухожильных рефлексов.
Развивающиеся первоначально атрофии затрагивают в первую очередь абдукторы и разгибатели стопы. Результатом является свисание стопы, невозможность ходьбы на пятках и своеобразная походка, напоминающая вышагивание лошади, — степпаж. Далее поражаются приводящие мышцы и сгибатели стопы. Тотальная атрофия мышц стопы приводит к ее деформации с высоким сводом, по типу стопы Фридрейха; формируются молоткообразные пальцы стопы. Постепенно атрофический процесс переходит на более проксимальные отделы ног — голени и нижние части бедер. В результате атрофии мышц голени возникает болтающаяся стопа. Из-за атрофии дистальных отделов ног при сохранности мышечной массы проксимальных отделов ноги приобретают форму перевернутых бутылок.
Зачастую при дальнейшем прогрессировании болезни Шарко-Мари-Тута атрофии появляются в мышцах дистальных отделов рук — вначале в кистях, а затем и в предплечьях. Из-за атрофии гипотенара и тенара кисть становиться похожей на обезьянью лапу. Атрофический процесс никогда не затрагивает мышцы шеи, туловища и плечевого пояса.
Часто невральная амиотрофия Шарко-Мари-Тута сопровождается легкими фасцикулярными подергиваниями мышц рук и ног. Возможна компенсаторная гипертрофия мышц проксимальных отделов конечностей.
Сенсорные нарушения при невральной амиотрофии характеризуются тотальной гипестезией, однако поверхностная чувствительность (температурная и болевая) страдает значительно больше глубокой. В некоторых случаях наблюдается цианоз и отек кожи пораженных конечностей.
Тремор рук.
Диагностика
Возраст начала заболевания, его типичная клиника, симметричный характер поражения, медленное неуклонное распространение атрофий и усугубляющаяся в связи с этим симптоматика во многих случаях позволяют предположить невральную амиотрофию. Проводимый неврологом осмотр выявляет мышечную слабость в стопах и голенях, деформацию стоп, отсутствие или значительное снижение ахилловых и коленных рефлексов, гипестезию стоп. Для дифференциации ШМТ от других нервно-мышечных заболеваний (миотонии, миопатии, БАС, невропатией) проводится электромиография и электронейрография. С целью исключения метаболической невропатии проводится определение сахара крови, исследование гормонов щитовидной железы, тест на наркотики.
Всем пациентам для уточнения диагноза рекомендована консультация генетика и проведение ДНК-диагностики. Последняя не дает 100% точного результата, т. Пока известны не все генетические маркеры ШМТ. Более точным способом диагностики является введенное в 2010г. Секвенирование генома. Однако это исследование пока является слишком дорогостоящим для широкого использования.
Иногда возникают затруднения в дифференциальной диагностике болезни Шарко-Мари-Тута с невритом Дежерина – Сотта, дистальной миопатией Гоффманна, хроническими полиневропатиями. В таких случаях может потребоваться биопсия мышц и нервов.
Лечение
На современном этапе радикальные способы лечения генных болезней не разработаны. В связи с этим применяется симптоматическая терапия. Проводятся повторные курсы внутримышечного введения витаминов группы В и витамина Е. С целью улучшения мышечной трофики используют АТФ, инозин, кокарбоксилазу, глюкозу. Назначаются ингибиторы холинэстеразы (неостигмин, оксазил, галантамин), препараты для улучшения микроциркуляции и антиоксиданты (никотиновая кислота, пентоксифиллин, мельдоний).
Наряду с фармакотерапией по рекомендации физиотерапевта активно применяются физиотерапевтические методики: электрофорез, СМТ, электростимуляция, диадинамотерапия, грязелечение, УЗ-терапия, оксигенобаротерапия. Целесообразно водолечение сероводородными, сульфидными, хвойными, радоновыми лечебными ваннами. Большое значение в сохранении двигательной активности пациентов, предотвращении развития деформаций и контрактур имеют ЛФК и массаж. При необходимости ортопедом назначается ортопедическое лечение.
Источник