Болезнь хагемана код мкб

Содержание
- Синонимы диагноза
- Описание
- Симптомы
- Причины
- Лечение
Другие названия и синонимы
Синдром марионетки, Синдром Петрушки, Синдром счастливой куклы.
Названия
Синдром Ангельмана.
Дефект в 15-й хромосоме — Причины Синдром Ангельмана
Синонимы диагноза
Синдром марионетки, Синдром Петрушки, Синдром счастливой куклы.
Описание
Cиндром Ангельмана является генетическим заболеванием, которое вызывает отклонения в развитии пациента, неврологические проблемы в виде нарушения речи, трудности при ходьбе и, в некоторых случаях, судороги.
Синдром Ангельмана обычно не обнаруживается, пока родители не начинают замечать задержку в развитии ребенка в возрасте 6-12 месяцев. Явные дефекты проявляются в возрасте 2 — 3 лет.
Как правило, синдром Ангельмана не влияет на продолжительность жизни.
Симптомы
Признаки синдрома Ангельмана включают в себя:
1. Задержка в развитии ребенка.
2. Отсутствие или минимальный объем речи.
3. Неспособность ходить, нарушение равновесия (атаксия).
4. Тремор при движении рук и ног.
5. Ребенок часто улыбается и смеется.
6. Повышенная возбудимость личности.
Люди с синдромом Ангельмана может также иметь другие признаки и симптомы, в том числе:
1. Судороги, которые обычно начинаются с 2 — 3 лет.
2. Резкость движений.
3. Уменьшенный размер головы, скошенный затылок (микробрахицефалия).
4. Косоглазие.
5. Точкообразные движения языка.
6. Гипопигментации волос, кожи и радужки глаз.
Большинство детей с синдромом Ангельмана не проявляют признаков расстройства при рождении. Первые симптомы в виде задержки в развитии проявляются в возрасте 6 — 12 месяцев.
Миоклония. Судороги. Тремор.
Причины
Синдром Ангельмана является генетическим расстройством. Чаще всего он связан с проблемами гена, расположенного в 15-ой хромосоме (ген UBE3A).
Обычно, только материнская копия гена UBE3A активна в мозге. Большинство случаев синдрома Ангельмана развивается, когда часть материнской 15-й хромосомы, содержащая этот ген, отсутствует или повреждена. В небольшом числе случаев, этот синдром развивается, когда наследуются две отцовские копии, вместо одной отцовской и одной материнской копии (отцовская дисомия).
Синдром Ангельмана встречается редко. В большинстве случаев исследователи не знают, что вызывает генетические изменения, которые приводят к синдрому Ангельмана. Большинство людей с этой патологией не имеют наследственной истории этого заболевания. В небольшом проценте случаев, однако, синдром Ангельмана может быть унаследован от родителей.
Лечение
Так как нет способов восстановления хромосомных дефектов, нет и этиотропного лечения для синдрома Ангельмана.
В таких случаях проводится симптоматическое лечение, обусловленное проявлениями синдрома. В зависимости от симптомов, лечение синдрома Ангельмана может включать в себя следующие пункты:
1. Антиконвульсанты. Лекарства могут быть необходимы для контроля припадков, вызванных синдромом Ангельмана.
2. Лечебная физкультура. Дети с синдромом Ангельмана могут научиться ходить лучше и преодолеть другие проблемы с движениями с помощью физиотерапии.
3. Хотя люди с синдромом Ангельмана обычно не овладевают вербальным языком в полной мере, общение очень полезно для таких пациентов. Невербальные языковые навыки могут быть разработаны на основе языка жестов.
4. Поведенческая терапия. Поведенческая терапия может помочь детям с синдромом Ангельмана преодолеть гиперактивность, улучшить концентрацию внимания. Многие больные с синдромом Ангельмана способны строить отношения с друзьями и создать семью.
Источник
Исключены:
- лихорадка неясного происхождения (во время) (у):
- родов (O75.2)
- новорожденного (P81.9)
- лихорадка послеродового периода БДУ (O86.4)
Боль в области лица
Исключены:
- атипичная боль в области лица (G50.1)
- мигрень и другие синдромы головной боли (G43-G44)
- невралгия тройничного нерва (G50.0)
Включена: боль, которая не может быть отнесена к какому-либо определенному органу или части тела
Исключены:
- хронический болевой личностный синдром (F62.8)
- головная боль (R51)
- боль (в):
- животе (R10.-)
- спине (M54.9)
- молочной железе (N64.4)
- груди (R07.1-R07.4)
- ухе (H92.0)
- области таза (H57.1)
- суставе (M25.5)
- конечности (M79.6)
- поясничном отделе (M54.5)
- области таза и промежности (R10.2)
- психогенная (F45.4)
- плече (M25.5)
- позвоночнике (M54.-)
- горле (R07.0)
- языке (K14.6)
- зубная (K08.8)
- почечная колика (N23)
последние изменения: январь 2015
R53
Недомогание и утомляемость
Астения БДУ
Слабость:
- БДУ
- хроническая
Общее физическое истощение
Летаргия
Усталость
Исключены:
- слабость:
- врожденная (P96.9)
- старческая (R54)
- истощение и усталость (вследствие) (при):
- нервной демобилизации (F43.0)
- чрезмерного напряжения (T73.3)
- опасности (T73.2)
- теплового воздействия (T67.-)
- неврастении (F48.0)
- беременности (O26.8)
- старческой астении (R54)
- синдром усталости (F48.0)
- после перенесенного вирусного заболевания (G93.3)
последние изменения: январь 2012
Старческий возраст без упоминания о психозе
Старость без упоминания о психозе
Старческая:
- астения
- слабость
Исключен: старческий психоз (F03)
R55
Обморок [синкопе] и коллапс
Кратковременная потеря сознания и зрения
Потеря сознания
Исключены:
- нейроциркуляторная астения (F45.3)
- ортостатическая гипотензия (I95.1)
- неврогенная (G23.8)
- шок:
- БДУ (R57.9)
- кардиогенный (R57.0)
- осложняющий или сопровождающий:
- аборт, внематочную или молярную беременность (O00-O07, O08.3)
- роды и родоразрешение (O75.1)
- послеоперационный (T81.1)
- приступ Стокса-Адамса (I45.9)
- обморок:
- синокаротидный (G90.0)
- тепловой (T67.1)
- психогенный (F48.8)
- бессознательное состояние БДУ (R40.2)
последние изменения: январь 2016
Исключены: судороги и пароксизмальные приступы (при):
- диссоциативные (F44.5)
- эпилепсии (G40-G41)
- новорожденного (P90)
Исключены:
- шок (вызванный):
- анестезией (T88.2)
- анафилактический (вследствие):
- БДУ (T78.2)
- неблагоприятной реакции на пищевые продукты (T78.0)
- сывороточный (T80.5)
- осложняющий или сопровождающий аборт, внематочную или молярную беременность (O00-O07, O08.3)
- воздействием электрического тока (T75.4)
- в результате поражения молнией (T75.0)
- акушерский (O75.1)
- послеоперационный (T81.1)
- психический (F43.0)
- травматический (T79.4)
- синдром токсического шока (A48.3)
последние изменения: январь 2015
R58
Кровотечение, не классифицированное в других рубриках
Кровотечение БДУ
Включены: опухшие железы
Исключены: лимфаденит:
- БДУ (I88.9)
- острый (L04.-)
- хронический (I88.1)
- мезентериальный (острый) (хронический) (I88.0)
Исключена: задержка полового созревания (E30.0)
Исключены:
- булимия БДУ (F50.2)
- расстройства приема пищи неорганического происхождения (F50.-)
- недостаточность питания (E40-E46)
Исключены:
- синдром истощения как результат заболевания, вызванного ВИЧ (B22.2)
- злокачественная кахексия (C80.-)
- алиментарный маразм (E41)
последние изменения: январь 2010
Эта категория не должна использоваться в первичном кодировании. Категория предназначена для использования в множественном кодировании, чтобы определить данный синдром, возникший по любой причине. Первым должен быть присвоен код из другой главы, чтобы указать причину или основное заболевание.
добавлено: январь 2010
R69
Неизвестные и неуточненные причины заболевания
Болезненность БДУ
Недиагностированная болезнь без уточнения локализации или пораженной системы
Источник
Связанные заболевания и их лечение
Описания заболеваний
Национальные рекомендации по лечению
Содержание
- Синонимы диагноза
- Описание
- Симптомы
- Причины
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Другие названия и синонимы
Бронзовый диабет, Пигментный цирроз.
Названия
Гемохроматоз.
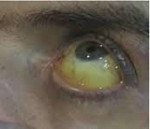
Гемохроматоз
Синонимы диагноза
Бронзовый диабет, Пигментный цирроз.
Описание
Гемохроматоз — наследственное заболевание, передаваемое по аутосомно-рецессивному типу, характеризующееся нарушением кишечной абсорбции железа с накоплением его в клетках паренхиматозных органов: печени, поджелудочной железы и сердца. Так как печень является основным органом депонирования железа в организме, поражение печени, проявляющееся микронодулярним циррозом, является общим и ранним признаком наследственного гемохроматоза. Может развиваться застойная сердечная недостаточность, вторично ведущая к дилатационной кардиомиопатии со склонностью к накоплению железа в мышце сердца. Эндокринные признаки наследственного гемохроматоза характеризуются гипогонадизмом, часто в комбинации с частичной дисфункцией переднего гипофиза, первичной тестикулярной недостаточностью, сахарным диабетом. Также характерны артропатии и патологическая пигментация кожи.
Накопление железа в поджелудочной железе, сопровождается ее циррозом и снижением экзо- и эндокринной функций. Поражение поджелудочной железы при гемохроматозе сопровождается сахарным диабетом, а учитывая темную пигментацию кожи, гемохроматоз так же называют бронзовым диабетом.
У больных идиопатическим гемохроматозом повышена абсорбция железа в кишечнике при нормальном его содержании в пище. Повышенное депонирование железа в клетках печени и других внутренних органах ведет к дегенерации клеток и фиброзу в сочетании с очень незначительной воспалительной реакцией. В норме общие запасы железа в организме не превышают 4-5 г, ежедневная потребность в железе составляет у мужчин 1 мг и у менструирующих женщин 1,5 мг. Избыточное всасывание железа при гемохроматозе может приводить к увеличению количества депонируемого железа до 20-60 г. Концентрация железа в печени (в норме 7-100 мкг на 100 мг сухой массы) у всех больных гемохроматозом превышает 1000 мкг на 100 мг (т. Е. 1% сухой массы печени). Первоначально железо откладывается в клетках печени в форме легко мобилизуемого водорастворимого, содержащего 23% железа, белка ферритина, позднее — в форме окруженных однослойной мембраной гранул белка гемосидерина, содержащих 35% железа. Гемосидерин с трудом мобилизуется, состоит из полимеризованных молекул ферритина и дает положительную реакцию с берлинской лазурью (реакция Перлса).
Гемохроматоз
Симптомы
Классическая триада развернутой стадии гемохроматоза: сероватая с голубоватым оттенком пигментация кожи, главным образом в области конечностей, лица, шеи и половых органов (меланодермия), скудное оволосение, сахарный диабет и гепатомегалия (бронзовый диабет, пигментный цирроз печени) наблюдается более чем у 80% больных. Печень у больных гемохроматозом равномерно увеличена, плотная, гладкая и нередко болезненная. Увеличение селезенки наблюдается на более поздних стадиях заболевания. Конечная стадия заболевания — макронодулярный цирроз печени. Сахарный диабет часто является инсулинозависимым, а у некоторых больных может отмечаться инсулинорезистентность. Возможен и латентный диабет, выявляемый пробой с сахарной нагрузкой. У 20% больных отмечается пигментация не только кожи, но и слизистых оболочек. У 7-19% больных гемохроматозом развивается гепатоцеллюлярный рак печени. Часто наблюдается цирроз поджелудочной железы, чем объясняется развитие при гемохроматозе инсулинзависимого сахарного диабета.
Боль в голеностопе. Гипербилирубинемия.
Причины
Заболевание обусловлено, по-видимому, генетическим ферментативным дефектом, природа которого еще не выяснена. Оно передается аутосомально-рецессивно и ассоциировано с некоторыми антигенами тканевой совместимости, среди которых преобладают HLA-A3 и HLA-B7. Частота гена идиопатического гемохроматоза в общей популяции составляет 5,6%, что соответствует частоте заболевания 0,3%.
Мужчины болеют в 10 раз чаще, чем женщины, обычно в возрасте 40-60 лет. В начальной стадии, медленно прогрессирующей в цирроз печени, на протяжении ряда лет преобладают жалобы на выраженную слабость, быструю утомляемость, потерю в массе тела, снижение половой функции у мужчин. Часто отмечаются боль в правом подреберье, суставах в связи с хондрокальцинозом крупных суставов, сухость и атрофические изменения кожи, атрофия яичек. Поражение миокарда с сердечной недостаточностью и аритмией может быть начальным проявлением болезни.
Лечение
Наиболее эффективным методом лечения идиопатического гемохроматоза являются систематические кровопускания под контролем самочувствия больного, картины красной крови, показателей обмена железа и повторных пункций печени. Первоначально при каждой венопункции удаляют по 500 мл крови. Кровопускания повторяются с недельными интервалами до развития умеренной анемии. Далее интервалы постепенно увеличивают до 3 мес, продолжая лечение до полного исчезновения избыточного депонирования железа в печени. Благодаря этому удается снизить летальность больных в течение 5 лет с 67 до 11%.
При данном виде цирроза совершенно необходимо назначение антиоксидантов, так как и при гемохроматозе, инициируется образование чрезмерного количества свободных радикалов кислорода, водорода, продуктов перекисного окисления липидов, которые непосредственно снижают резистентность гепатоцитов и, как следствие, функциональную активность печени, приводя и усугубляя гепатопривную недостаточность. Кроме того, продукты пероксидации липидов активируют звездчатые ретикулоэндотелиоциты и липоциты (в основном это касается малонового диальдегида) и как результат интенсифицируются иммунное воспаление и коллагенообразование в печени. Назначают ессенциале, ливолин, легалон, симепар по 2 капс. 3 раза в день 3-4 мес, а также препараты, содержащие витамины Е (альвитил, генсамин, форматон, триовит, токоферол) и цинк (намацит). Последние назначают в среднетерапевтических дозах в течение 30 дней.
Основные медуслуги по стандартам лечения | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Клиники для лечения с лучшими ценами
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Источник