Болезнь фридриха код мкб

Расстройства координации движений встречаются при различных заболеваниях ЦНС. Все они связаны с нарушениями в структуре мозжечка. Чаще всего такие патологии носят генетический характер и передаются по наследству. Одной из подобных аномалий является болезнь Фридрейха. Эта патология считается довольно распространенной по сравнению с другими хромосомными дефектами. Помимо нервной системы, она распространяется и на другие органы. В первую очередь это касается сердца и мышц. По сравнению с другими атаксиями, данное заболевание нельзя диагностировать в раннем возрасте, так как оно дает о себе знать только к 20 годам.
Патология Фридрейха – что это такое?
Болезнь Фридрейха – это неврологическое нарушение, которое относится к генетическим патологиям. Заранее заподозрить данную аномалию очень сложно, так как она никак не проявляет себя в детском возрасте. Заболевание часто носит семейный характер. Поэтому при таких нарушениях беременным женщинам необходимо проходить полное генетическое обследование.
История и распространение патологии
Атаксия Фридрейха была впервые выделена как самостоятельное заболевание в 60-х годах XIX века. Тогда же обнаружено, что эпидемиология данной аномалии неравномерна. Также можно проследить связь между заболеванием и этнической принадлежностью пациентов. Чаще всего данная патология встречается у людей, рожденных от кровных браков. По этой же причине она больше распространена среди небольших этносов. По сравнению с другими атаксиями, болезнь Фридрейха является довольно распространенной. Она встречается у 1–7 людей на 100 тысяч населения. В настоящее время уже известно, какие генетические нарушения приводят к данной патологии, поэтому при отягощенном наследственном анамнезе ее можно диагностировать на этапе внутриутробного развития плода.

Причины болезни Фридрейха
Чтобы понять, откуда берется болезнь Фридрейха, как передается и распространяется, необходимо знать этиологию этой аномалии. Данная патология относится к наследственным хромосомным дефектам. Она передается по аутосомно-рецессивному признаку через поколения. Поэтому болезнь часто можно обнаружить у нескольких членов семьи. При зачатии детей от родственных браков частота появления данной патологии увеличивается. Поэтому таким семьям необходимо более тщательное обследование при планировании ребенка. Ученые установили, что у всех пациентов с данным заболеванием имеется генетический дефект в девятой хромосоме. Если у одного из родителей обнаружена такая аномалия, то шанс, что ребенок будет болен, составляет 50%. Несмотря на наследственный характер заболевания, существуют провоцирующие факторы окружающей среды, способные вызывать мутации хромосом во время эмбриогенеза. К ним относятся: вредные привычки (наркомания, алкоголизм), радиация, стрессовые ситуации.
Патогенез аномалии Фридрейха
Основным проявлением заболевания является мозжечковая атаксия. Она возникает из-за постепенной дегенерации клеток нервной системы. Механизм повреждения заключается в том, что из-за мутантного гена, содержащегося в 9-й хромосоме, в организме перестает вырабатываться вещество – фратаксин. Вследствие этого клетки накапливают повышенное количество железа. Это запускает следующий патологический процесс – накопление свободных радикалов и перекисное окисление липидов. В результате происходит разрушение клеточных оболочек и нервной ткани. Чаще всего этот процесс поражает задние рога спинного мозга и пирамидную систему. Эти структуры отвечают за двигательную функцию организма, поэтому происходит расстройство координации. В некоторых случаях поражаются и другие структуры мозга: передние рога, периферические волокна, ЦНС. Чаще всего дегенерация не охватывает черепно-мозговые нервы и жизненно важные центры (дыхательный, сосудистый). В редких случаях наблюдаются расстройства зрения и слуха.
Помимо неврологической симптоматики, происходят изменения костной системы. Часто у больных с аномалией Фридрейха наблюдается искривление позвоночника и деформация стоп. Помимо этого, патологический процесс захватывает сердечную мышцу. При этом нормальные кардиомиоциты заменяются фиброзной и жировой тканью.
Клиническая картина заболевания
Исходя из патогенеза заболевания, клиническая картина зависит от степени распространенности поражения структур спинного и головного мозга. Типичным синдромом является атаксия. Но в некоторых случаях наблюдаются и другие нарушения. Если диагностирована болезнь Фридрейха, симптомы патологии выделяют следующие:
- Мозжечковая атаксия. Этот синдром появляется у больных примерно к 20 годам. Основные его проявления – изменение походки, шаткость, нистагм. При неврологическом обследовании пациенты не могут выполнить пяточно-коленную пробу и неустойчивы в положении Ромберга.
- Мышечная гипотония. Наряду с изменением походки, наблюдается прогрессирующая слабость в ногах. Позже процесс переходит и на мышцы верхнего плечевого пояса. Пациентам сложно удерживать конечности в определенном положении. Это приводит к слабости суставов и патологическому разгибанию конечностей («стопа Фридрейха»). У некоторых больных наблюдается обратное явление – спазм мускулатуры, парезы.
- Гиперкинезы. Из-за поражения пирамидной системы головного мозга у пациентов с болезнью Фридрейха наблюдается тремор головы и конечностей. Иногда возможны нарушения мимики – тики.
- Нарушения костно-суставной системы. У больных часто обнаруживается сколиоз и другие искривления позвоночника. Из-за «разболтанности» суставов нижних конечностей происходит характерная для данной патологии деформация стопы. При этом свод ее углубляется, а проксимальные фаланги выворачиваются.
- Постепенное снижение сухожильных рефлексов.
- Изменение почерка.
- Гипертрофическая кардиомиопатия. Этот синдром наблюдается практически у всех больных (90% случаев). Клинически он проявляется глухостью сердечных тонов при аускультации, увеличением размеров сердца и появлением систолического шума. При этом человек жалуется на боли в грудной области, одышку.
Менее типичными симптомами является нарушение чувствительности, офтальмоплегия, птоз. Иногда дегенеративные процессы захватывают слуховой и зрительный нерв. При этом происходит снижение слуха, слепота.
Диагностические критерии атаксии Фридрейха
Заподозрить болезнь Фридрейха не составляет большого труда для опытного невролога. В первую очередь доктор обращает внимание то, что нарушения начинаются в определенном возрасте. Обычно в детстве и подростковом периоде жалобы полностью отсутствуют. Помимо этого, в ходе сбора анамнеза часто наблюдается связь между заболеванием и наследственными факторами (браки среди родственников, атаксия у кого-то из членов семьи). Если обнаружена болезнь Фридрейха, диагностика у невролога будет проходить по следующей схеме (моменты, на которые обращает внимание врач):
- Мышечная слабость (чаще на нижних конечностях).
- Снижение сухожильных рефлексов.
- Шаткое положение в позе Ромберга.
- Горизонтальный нистагм.
- Невозможность выполнить пяточно-коленную пробу.
- Парезы и параличи (встречаются редко).
- Гиперкинезы.
Помимо этого, требуется осмотр хирурга. Врач определяет нарушения костно-суставной системы нижних конечностей, искривление позвоночного столба. При подозрении на данную патологию необходим также осмотр офтальмолога и кардиолога.
Основным методом диагностики является генетический анализ, в ходе которого обнаруживают хромосомную аномалию. Также проводят МРТ головного мозга.
Болезнь Фридрейха: код по МКБ 10
Несмотря на клинический диагноз, каждую патологию необходимо регистрировать по международной номенклатуре. Болезнь Фридрейха по МКБ-10 имеет код G11.1. Это означает, что, согласно международной шкале, данная патология имеет следующее название: ранняя мозжечковая атаксия.
Болезнь Фридрейха: лечение патологии
Несмотря на то что неврология как наука в настоящее время довольно хорошо развита, данное заболевание невозможно полностью вылечить. Это связано с тем, что его причиной является генетическая мутация, на которую нельзя воздействовать. Тем не менее врачи проводят коррекцию неврологических нарушений, облегчая жизнь пациентам. Используются антиоксиданты (медикамент «Мексидол»), средства для улучшения мозгового кровообращения (препараты «Пирацетам», «Церебролизин»). Эти лекарственные вещества позволяют замедлить дегенеративные процессы. Для улучшения двигательной функции проводится физиотерапия, ортопедическая коррекция, массаж. В некоторых случаях необходимо хирургическое лечение (при значительном искривлении стоп).
Профилактика заболевания и осложнений
К сожалению, невозможно заранее предвидеть развитие болезни Фридрейха. Тем не менее патология не относится к очень редким аномалиям. Поэтому при отягощенном наследственном анамнезе у беременной женщины необходимо провести генетический анализ плода.
Чтобы предупредить развитие осложнений у больных с патологией Фридрейха, необходим постоянный контроль врачей. Несмотря на то что нарушение относится к неврологическим проблемам, также важно наблюдение хирурга, кардиолога, окулиста и генетика. Чтобы избежать быстрого прогрессирования болезни, проводится поддерживающая терапия. Также пациентам необходим специальный уход и поддержка близких людей.

Прогноз при заболевании Фридрейха
Учитывая то, что патология относится к медленно прогрессирующим хромосомным аномалиям, пациенты доживают в среднем до 30–40 лет. Чаще всего неврологические нарушения не затрагивают умственные способности и жизненно важные структуры мозга. Тем не менее больным требуется постоянное наблюдение врачей. Также пациенты должны заниматься лечебной физкультурой для поддержания мышечного тонуса. Чаще всего причиной смерти становятся нарушения сердечно-сосудистой системы. Гипертрофическая кардиомиопатия с годами приводит к застойной СН, итогом которой становится болезнь Фридрейха. Фото пациентов с данной патологией можно увидеть в данной статье и специальной литературе.
Источник
Содержание
- Описание
- Симптомы
- Причины
- Лечение
Названия
Семейная атаксия Фридрейха.
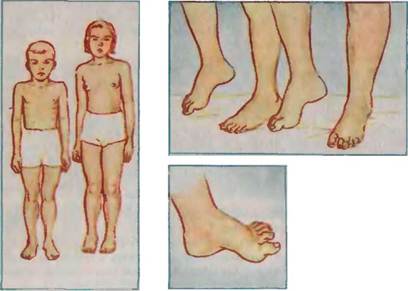
Сибсы с семенной атаксиеи Фридрейха
Описание
Семейная атаксия Фридрейха — это хроническое прогрессирующее заболевание, основным клиническим проявлением которого служит атаксия, обусловленная главным образом комбинированным поражением спинальных систем.
Симптомы
Основным симптомом болезни является атаксия. Походка больных была обозначена Шарко как табетически-мозжечковая. Больные ходят, широко расставляя ноги, отклоняясь от прямого направления в обе стороны; походка неуверенная, неуклюжая. Наблюдается также статическая атаксия; часто положителен симптом Ромберга. По мере развития заболевания нарушения координации распространяются на руки, мышцы грудной клетки, лицо. Нарушается почерк, могут наблюдаться своеобразные атак-тические нарушения дыхания, изменяется мимика. Речь становится медленной, немодулированной, толчкообразной. У больных могут отмечаться дизметрия, адиадохокинез, различные гиперкинезы, как правило, сопровождающие активные движения. Мышечный тонус понижен. В поздних стадиях заболевания может развиваться нижний спастический парапарез.
Характерным и ранним признаком болезни является отсутствие или снижение сухожильных и надкостничных рефлексов. В первую очередь сухожильные рефлексы угасают на ногах, затем арефлексия распространяется и на верхние конечности. С развитием спастического парапареза сухожильные рефлексы-могут появляться вновь. Часто, особенно в поздних стадиях болезни, вызываются патологический рефлекс Бабинского, защитные рефлексы. Характерно снижение глубокой чувствительности. Поверхностные виды чувствительности обычно не нарушаются. К характерным признакам болезни относится крупноразмашистый нистагм. При отоневрологическом обследовании в большинстве случаев выявляется двусторонняя вестибулярная арефлексия или асимметрия рефлекторного нистагма. У ряда больных отмечено снижение слуха. Атрофия зрительного нерва и поражение глазодвигательных нервов в отличие от мозжечковой атаксии встречаются редко. В большинстве случаев интеллект сохранен. Однако могут наблюдаться различные степени олигофрении. Отмечаются изменения ЭЭГ, которые выражаются в нарушении альфа- и бета-ритма, наличии нерегулярных острых волн и групп медленных колебаний.
Атаксии Фридреиха свойственны различные экстраневральные нарушения; наиболее частыми из них являются изменения скелета. И поражение сердца. Первые выражаются в кифосколиозе и характерном изменении формы стопы: увеличение свода и экстензия пальцев, главным образом I пальца в основной фаланге; имеется наклонность к частым вывихам суставов.
Проявлениями поражения сердца служат тахикардия, приступообразные боли в области сердца, одышка при физическом напряжении, расширение границ сердца, систолический шум. При электрокардиографическом исследовании обнаруживаются нарушения ритма, изменения предсердно-желудочковой и внутрижелудочковои проводимости, деформация предсердного зубца. У больных нередко отмечаются врожденные пороки сердца.
В ряде случаев атаксия Фридреиха сочетается с диабетом, который при этой форме наследственной атаксии встречается чаще, чем в общей популяции. Из других эндокринных нарушений могут наблюдаться инфантилизм и гипогонадизм. Имеются единичные наблюдения сочетания атаксии Фридреиха с врожденной катарактой.
У клинически здоровых родственников больных атаксией Фридреиха часто обнаруживаются отдельные признаки, свойственные болезни. Наиболее частыми из них являются нистагм и снижение или отсутствие сухожильных рефлексов. В ряде случаев отдельным семьям бывают свойственны определенные аномалии. По всей вероятности, их следует рассматривать как фенотипические проявления гетерозиготного носительства патологического гена. Необходимо учитывать, однако, что в детском возрасте эти признаки могут быть первыми проявлениями развивающегося заболевания.
Средний возраст начала болезни — 13 лет. Заболевание медленно, но неуклонно прогрессирует. Средняя продолжительность болезни де-зависимо от возраста ее начала-16 лет. Различные инфекции и другие экзогенные вредности могут способствовать развитию заболевания и ухудшать его течение.
В некоторых семьях атаксия Фридреиха протекает атипично: наряду с атаксией могут иметь место симптомы, свойственные мозжечковой атаксии, семейной спастической параплегии, невральной амио-трофии. Данные случаи рассматриваются как промежуточные формы между этими заболеваниями; предполагается, что они вызываются самостоятельными генами.
Тремор. Тремор рук.
Причины
Заболевание является наследственным и передается по аутосомно-рецессивному типу. Среди родителей больных отмечена повышенная частота кровных браков. Доминантное наследование установлено лишь в единичных семьях. Не исключена возможность, что эти случаи представляют собой атипичные формы других гередитарных атаксий.
Характерным патологоанатомическим признаком атаксии Фридрейха является дегенерация задних и боковых столбов спинного мозга. Пучки Голля поражаются в большей степени, чем пучки Бурдаха. Страдают клетки столбов Кларка и начинающийся от них задний спинально-мозжечковый путь. Поражение пирамидных путей обычно начинается с поясничного отдела. Дегенерация спинальных путей обычно прослеживается до продолговатого мозга.
Лечение
Лечение симптоматическое. Применяется специальная система лечебной гимнастики, направленная преимущественно на уменьшение координаторных нарушений. При назначении упражнений необходимо учитывать возможность кардиальной патологии. При наличии последней назначают соответствующую терапию. Показаны и общеукрепляющие средства.
Источник
Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Дифференциальная диагностика
- Лечение
- Прогноз
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Болезнь Норри.
Болезнь Норри
Описание
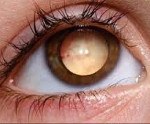
Болезнь Норри. Генетическое заболевание, характеризующееся появлением псевдоглиомы сетчатки обоих глаз в первые месяцы жизни ребенка и другими нарушениями и пороками развития. Симптомами патологии являются полная слепота, наличие гиперплазии сетчатки и пигментного эпителия радужки, в ряде случаев – прогрессирующая умственная отсталость и нарушения слуха. Диагностика производится с помощью офтальмологических исследований, генетических анализов и изучения наследственного анамнеза пациента. Специфического лечения болезни Норри не существует, работа с больным психологов и невропатологов может задержать развитие или уменьшить выраженность умственной неполноценности.
Дополнительные факты
Болезнь Норри (окулоцереброакустический синдром, врожденная двусторонняя псевдоглиома сетчатки) – редкое наследственное заболевание, представляющее собой врожденную ретинальную дисплазию, сопровождающуюся патологией слуха и ЦНС. Впервые было описано в 1926 году датским врачом-офтальмологом Г. Норри и почти в то же время – его соотечественником М. Варбургом. Представляет собой генетическое заболевание, наследующееся по рецессивному, сцепленному с Х-хромосомой механизму. Поэтому болезнь Норри наблюдается только у мальчиков, хотя имеются противоречивые данные о наличии нарушений зрения и у женщин-носительниц, представляющих собой гетерозиготы. Ввиду значительной редкости заболевания его популяционная частота до сих пор не выяснена.
Болезнь Норри
Причины
Причиной развития заболевания является мутация гена NDP, расположенного на Х-хромосоме. Он кодирует особый белок, названный фактором болезни Норри (Norrie Disease Protein), его функции в человеческом организме досконально не изучены. В процессе исследования лабораторных животных, в частности мышей, было выявлено, что подобный протеин, более чем на 90% идентичный вышеуказанному фактору, контролирует дифференцировку клеток сетчатки. Кроме того, он участвует в формировании межклеточных связей в центральной нервной системе. Вполне возможно, что этот белок играет аналогичную роль в человеческом организме, именно поэтому нарушения его структуры вызывают поражение сетчатки и центральной нервной системы, что и наблюдается при болезни Норри.
Патогенез нарушений при этой патологии также не изучен, имеется лишь несколько основных гипотез, пытающихся объяснить процессы, приводящие к развитию псевдоглиом сетчатки, гиперплазии реснитчатого тела и радужной оболочки. Предполагается нарушение работы рецепторов к ангиогенным факторам роста в сосудах сетчатки и других элементах нейроэктодермы. Вариабельность таких симптомов болезни Норри, как умственная отсталость и тугоухость предположительно объясняется большим количеством типов мутаций гена NDP – на сегодняшний день их известно более 30-ти. Методами современной генетики ученые пытаются отожествлять определенный характер течения патологии с конкретным типом или группой мутаций, но эти исследования тормозятся по причине редкости заболевания.
Симптомы
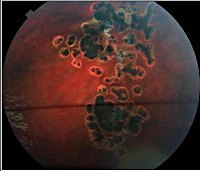
Проявления заболевания могут выявляться в разные сроки – на этапе внутриутробного развития (при помощи эхографии), сразу после рождения, в первые месяцы жизни человека. Статистически чаще встречается именно последний вариант обнаружения болезни Норри – как правило, родители обращаются к врачу по поводу двухсторонней лейкокории у их ребенка. Она обусловлена развитием дисплазии сетчатки с появлением псевдоглиальных разрастаний в виде бесформенных масс. Если осмотр глазного дна производится ранее их образования, то наблюдается складчатость сетчатой оболочки глаза, иногда ее отслойка. Эти проявления также можно обнаружить при осмотре глазного дна новорожденного с болезнью Норри, но возможно и их отсутствие в столь раннем возрасте. Очень редко симптомы заболевания выявляются у недоношенных детей или при профилактической эхографии – обнаруживается микрофтальм, фтизис одного или обоих глазных яблок, что можно увидеть при УЗИ глаза.
Впоследствии, в течение нескольких месяцев после появления псевдоглиальных диспластичных масс, развивается полная слепота, у большинства больных это происходит к 8-му месяцу жизни. Болезнь Норри также приводит к развитию катаракты и отеку роговицы – в редких случаях эти симптомы проявляются раньше лейкокории, поэтому изменения сетчатки становятся не видны. Конечным итогом офтальмологических изменений является атрофия и фтизис обоих глаз. Иногда патологические процессы в глазах приводят к выраженному болевому синдрому и делают ребенка беспокойным – это может служить косвенным признаком болезни.
Диагностика
Диагностика болезни Норри может быть как пренатальной, так и производиться после рождения методиками клинической офтальмологии, генетическими исследованиями и изучением наследственного анамнеза. При пренатальной диагностике на поздних сроках внутриутробного развития эхографическими методами можно выявить микрофтальм, отслоение сетчатки и другие нарушения формирования зрительного аппарата. Но это возможно не всегда, так как в большинстве случаев выраженные офтальмологические симптомы возникают уже после рождения. Намного более специфичным методом пренатальной диагностики болезни Норри будет выявление врачом-генетиком мутаций гена NDP в материале, получаемом при помощи амниоцентеза или биопсии ворсин хориона.
Офтальмологический осмотр глазного дна (офтальмоскопия) на различных этапах развития заболевания показывает наличие складок и отслоения клетчатки, возможны кровоизлияния, отек диска зрительного нерва. В дальнейшем эти изменения сменяются фиброваскулярными массами, соответствующими псевдоглиальной дисплазии. При проведении биопсии сетчатой оболочки глаза обнаруживается глиальная пролиферация, аналогичные процессы выявляются и в стекловидном теле. Конечными офтальмологическими симптомами болезни Норри будут помутнение роговицы, неоваскуляризация радужной оболочки и атрофия глаза.
Генетическая диагностика заболевания производится путем прямого секвенирования последовательности гена NDP с целью выявления мутаций. Объектом исследования могут выступать как собственно больные с подозрением на наличие болезни Норри, так и их ближайшие родственники, особенно мать – так как она может являться носительницей патологического гена. Именно генетический анализ является наиболее достоверным и однозначным в определении этого заболевания.
Дифференциальная диагностика
Дифференциальную диагностику следует проводить с ретинобластомой, синдромом Варбурга, синдромом Патау.
Лечение
Специфического лечения болезни Норри не существует, возможна только симптоматическая терапия. Однако и она не способна сохранить зрение больным или хотя бы замедлить прогрессирование патологии. В тех случаях, когда определяется выраженный болевой синдром, рекомендуют производить энуклеацию, или удаление глаза. При выявлении умственной отсталости, своевременно начатые занятия ребенка с психологом могут замедлить ее развитие, однако положительный результат такого лечения не гарантирован, так как степень слабоумия сильно зависит от генетических факторов. Нарушения слуха можно исправить ношением слухового аппарата в тех случаях, если умственная отсталость больного не достигает тяжелой степени.
Прогноз
В целом прогноз болезни Норри неблагоприятный – больные теряют зрение в раннем возрасте и в ряде случаев имеют выраженное слабоумие и нарушения слуха. В подростковом и взрослом возрасте возможны приступы острого психоза, нередко служащие поводом для госпитализации в специализированные учреждения. Профилактику болезни Норри можно производить путем пренатальной генетической диагностики на ранних сроках беременности. Особенно необходимо выполнять такую проверку в тех случаях, когда у родителей выявлено носительство дефектного гена NDP или имелись случаи болезни Норри у родственников.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник