Болезнь александера код мкб

Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 3 ноября 2016;
проверки требуют 13 правок.
Болезнь Александера — патологоанатомически демиелинизирующая лейкодистрофия — заболевание, проявляющееся в раннем детском возрасте (до года), является наследственным. Аутосомно-доминантный тип передачи. Свойственны нарушения метаболизма в астроцитах. Наблюдается задержка умственного развития, увеличение массы головного мозга, высокая температура, судорожные припадки, слабый мышечный тонус, прогрессирующая гидроцефалия, пирамидные знаки. У детей Болезнь Александера протекает катастрофически, иногда наступает смертельный исход при проявлениях спастической тетраплегии и децеребрационной ригидности. У взрослых более медленное течение заболевания, с ремиссиями и остановками. Прогрессирует заболевание быстро, рано наступает летальный исход.
Причины[править | править код]
В 95 % случаев болезнь Александера развивается в результате мутации в гене, расположенном на 17-й хромосоме. Обычно мутация возникает спонтанно, то есть родители являются совершенно здоровыми, их генотип не имеет подобных изменений.
Ген отвечает за продукцию глиального фибриллярного кислого белка GFAP. В случае мутации измененный белок GFAP накапливается во вспомогательных клетках нейронов (нейроглии), что препятствует обеспечению нейронов питательными веществами. Кроме того, при болезни Александера в самом измененном белке GFAP образуются узелковые образования, которые называют волокнами Розенталя. Последние мешают нормальному проведению нервных импульсов по миелиновым волокнам.
У 5 % людей, у которых диагностирована болезнь Александера, подобный или иной генетический дефект не обнаруживается, то есть причина развития остается неизвестной.
Симптомы[править | править код]
Заболевание впервые проявляет себя у людей в разном возрасте. В зависимости от этого принято выделять несколько клинических форм:
- инфантильная (младенческая);
- ювенильная (юношеская);
- взрослая.
Предполагается наличие так называемой неонатальной формы заболевания, когда ребенок рождается уже с проявлениями болезни. У таких детей обычно с первых дней жизни отмечается повышенное внутричерепное давление, аномально большой череп. Характерен судорожный синдром, выраженная задержка нервно-психического развития. К сожалению, продолжительность жизни таких детей не составляет и года. Некоторые ученые относят эту форму к инфантильной, но с очень ранним началом.
Инфантильная форма развивается в раннем детском возрасте, в среднем — по достижении 6 месяцев. У таких детей плохой аппетит, они часто срыгивают вплоть до рвоты. Отмечается патологически быстрое увеличение размеров головы, нарастание внутричерепного давления. Это сказывается на темпах физического и нервно-психического развития. Дети плохо прибавляют в весе, поздно начинают держать голову (после 3-х месяцев), садиться и ползать. По мере роста и развития ребенка развивается мышечная слабость в конечностях (парезы) наряду с повышенным мышечным тонусом (спастичность), что проявляется ограничением объема и силы произвольных движений. На фоне парезов в конечностях появляются непроизвольные движения: выкручивающие, червеобразные движения в пальцах рук, повороты головой с фиксацией позы и тому подобное. Эти явления называются гиперкинезами, в частности, хореоатетозом. Возможны судорожные эпилептические припадки. Страдает интеллект: дети не узнают близких, не проявляют интерес к игрушкам, они не овладевают навыками (например, не могут нанизать кольца на пирамидку в соответствующем возрасте). Также нарушается координация движений, наблюдаются подергивания глазных яблок (нистагм). Самостоятельная ходьба практически невозможна. Заболевание неуклонно прогрессирует и заканчивается смертью в течение 2-3 лет.
Ювенильная форма проявляет себя несколько позже, в возрасте от 4 до 14 лет, в среднем — около 9 лет. Хотя отдельные признаки заболевания могут появиться и раньше — в 2-3 года, но обычно их не связывают с болезнью Александера. Такие дети несколько отстают в нервно-психическом развитии, страдают от судорог. У них голова имеет больший размер по сравнению со сверстниками (но не настолько больший по сравнению с инфантильной формой). Несколько позже присоединяются нарушения речи (смазанность, нечеткость), поперхивание при приеме пищи, а затем и при глотании воды. Голос приобретает гнусавый оттенок. Движения языком затрудняются. Все эти изменения формируют бульбарные и псевдобульбарные расстройства, а возникают в результате поражения ствола мозга. По утрам больных беспокоят неукротимые рвоты. Так же, как и при инфантильной форме, появляется мышечная слабость в конечностях, которая постепенно нарастает.
Мышечный тонус увеличивается, мышцы становятся плотными и твердыми на ощупь, появляются патологические стопные признаки (симптом Бабинского и другие). Постепенно эти изменения охватывают все четыре конечности, что становится причиной расстройств передвижения и самообслуживания. Возможны нарушение равновесия, расстройства поведения. Обычно интеллектуальные расстройства выражены незначительно или вообще отсутствуют, хотя описаны случаи и резкого снижения мыслительных способностей. У больных с ювенильной формой периодически регистрируют рефлекторную остановку дыхания: апноэ. В конце концов, прогрессирующее поражение нервной системы заканчивается смертельным исходом, в среднем, через 10 лет от появления начальных клинических признаков заболевания.
Взрослая форма развивается в сроки от 20 до 70 лет. Клинические симптомы довольно разнообразны, поскольку могут быть отражением патологии любого участка головного мозга. Чаще всего это парезы и параличи с повышенным мышечным тонусом, нарушения координации движений и равновесия, непроизвольные неконтролируемые движения, нарушения речи и глотания. Снижение интеллекта незначительное. Часто выявляется нистагм и нарушение содружественных (одновременных и однонаправленных) движений глазными яблоками. Болезнь прогрессирует и неизбежно заканчивается летальным исходом (обычно от присоединения интекуррентных инфекций).
Диагностика[править | править код]
Диагностика заболевания прижизненно довольно затруднительна, потому что клинических симптомов, свойственных только болезни Александера, нет. И специфических изменений ни один из методов исследования не выявляет (не считая генетического анализа, который, тем не менее, необходимо еще назначить, подозревая эту болезнь).
При магнитно-резонансной томографии головного мозга (МРТ) при болезни Александера выявляется демиелинизация различных отделов мозга (при инфантильной и юношеской формах — преимущественно в лобных с распространением на другие области, при взрослой — более выражена в мозжечке и стволе мозга).
При электроэнцефалографии регистрируют изменения биоэлектрической активности мозга в лобных отделах.
Генетический анализ наиболее точно позволяет подтвердить диагноз болезни Александера: находят мутацию в гене GFAP на 17-й хромосоме (в 95 % случаев). Следует помнить, что у 5 % больных этим заболеванием генетический дефект не обнаружен и по сей день.
Подтверждением заболевания служит обнаружение волокон Розенталя (что возможно при биопсии мозга или уже после смерти при вскрытии).
Лечение[править | править код]
Сегодня медицина не располагает эффективными способами лечения болезни Александера. Возможно, будущее в этом направлении принадлежит генной инженерии.
После установления диагноза обычно проводят симптоматическую терапию, позволяющую облегчить и продлить жизнь больному:
- при парезах назначают стимуляторы нервно-мышечной проводимости (Нейромидин);
- при спастичности мышц — миорелаксанты (Баклофен, Сирдалуд, Мидокалм);
- при эпилептических припадках — противосудорожные препараты (Вальпроаты, Сибазон и другие);
- для уменьшения непроизвольных движений могут использоваться нейролептики (Галоперидол, Азалептин и другие).
Для передвижения используют специальные приспособления, в том числе и ортопедические. Пик болезни позволяет передвигаться только с помощью инвалидной коляски. Конечно, в терминальных стадиях заболевания больные нуждаются в постоянном постороннем уходе.
Болезнь Александера — редкое, в основном, генетически обусловленное заболевание. Его развернутая клиническая картина представляет собой двигательные, координаторные нарушения, проблемы с речью и приемом пищи. Почти все взрослые больные живут не более 10 лет с момента развития заболевания. Наиболее точным методом диагностики является генетический. Способы лечения находятся в стадии разработки, больным в настоящее время помогают только симптоматическими средствами.[1]
Примечания[править | править код]
Источник
Болезнь Александера — патологоанатомически демиелинизирующая лейкодистрофия — заболевание, проявляющееся в раннем детском возрасте (до года), является наследственным. Аутосомно-доминантный тип передачи. Свойственны нарушения метаболизма в астроцитах. Наблюдается задержка умственного развития, увеличение массы головного мозга, высокая температура, судорожные припадки, слабый мышечный тонус, прогрессирующая гидроцефалия, пирамидные знаки. У детей Болезнь Александера протекает катастрофически, иногда наступает смертельный исход при проявлениях спастической тетраплегии и децеребрационной ригидности. У взрослых более медленное течение заболевания, с ремиссиями и остановками. Прогрессирует заболевание быстро, рано наступает летальный исход.
Энциклопедичный YouTube
1/3
Просмотров:
1 971
2 561
366
✪ Техника Александера в театральной педагогике Харькова
✪ Как Техника Александера помогает решить проблему усталости после рисования
✪ Клиническое наблюдение. Случай тяжелого лечения болезни Гиппеля-Линдау у молодого пациента
Содержание
- 1 Причины
- 2 Симптомы
- 3 Диагностика
- 4 Лечение
- 5 Примечания
Причины
В 95 % случаев болезнь Александера развивается в результате мутации в гене, расположенном на 17-й хромосоме. Обычно мутация возникает спонтанно, то есть родители являются совершенно здоровыми, их генотип не имеет подобных изменений.
Ген отвечает за продукцию глиального фибриллярного кислого белка GFAP. В случае мутации измененный белок GFAP накапливается во вспомогательных клетках нейронов (нейроглии), что препятствует обеспечению нейронов питательными веществами. Кроме того, при болезни Александера в самом измененном белке GFAP образуются узелковые образования, которые называют волокнами Розенталя. Последние мешают нормальному проведению нервных импульсов по миелиновым волокнам.
У 5 % людей, у которых диагностирована болезнь Александера, подобный или иной генетический дефект не обнаруживается, то есть причина развития остается неизвестной.
Симптомы
Заболевание впервые проявляет себя у людей в разном возрасте. В зависимости от этого принято выделять несколько клинических форм:
- инфантильная (младенческая);
- ювенильная (юношеская);
- взрослая.
Предполагается наличие так называемой неонатальной формы заболевания, когда ребенок рождается уже с проявлениями болезни. У таких детей обычно с первых дней жизни отмечается повышенное внутричерепное давление, аномально большой череп. Характерен судорожный синдром, выраженная задержка нервно-психического развития. К сожалению, продолжительность жизни таких детей не составляет и года. Некоторые ученые относят эту форму к инфантильной, но с очень ранним началом.
Инфантильная форма развивается в раннем детском возрасте, в среднем — по достижении 6 месяцев. У таких детей плохой аппетит, они часто срыгивают вплоть до рвоты. Отмечается патологически быстрое увеличение размеров головы, нарастание внутричерепного давления. Это сказывается на темпах физического и нервно-психического развития. Дети плохо прибавляют в весе, поздно начинают держать голову (после 3-х месяцев), садиться и ползать. По мере роста и развития ребенка развивается мышечная слабость в конечностях (парезы) наряду с повышенным мышечным тонусом (спастичность), что проявляется ограничением объема и силы произвольных движений. На фоне парезов в конечностях появляются непроизвольные движения: выкручивающие, червеобразные движения в пальцах рук, повороты головой с фиксацией позы и тому подобное. Эти явления называются гиперкинезами, в частности, хореоатетозом. Возможны судорожные эпилептические припадки. Страдает интеллект: дети не узнают близких, не проявляют интерес к игрушкам, они не овладевают навыками (например, не могут нанизать кольца на пирамидку в соответствующем возрасте). Также нарушается координация движений, наблюдаются подергивания глазных яблок (нистагм). Самостоятельная ходьба практически невозможна. Заболевание неуклонно прогрессирует и заканчивается смертью в течение 2-3 лет.
Ювенильная форма проявляет себя несколько позже, в возрасте от 4 до 14 лет, в среднем — около 9 лет. Хотя отдельные признаки заболевания могут появиться и раньше — в 2-3 года, но обычно их не связывают с болезнью Александера. Такие дети несколько отстают в нервно-психическом развитии, страдают от судорог. У них голова имеет больший размер по сравнению со сверстниками (но не настолько больший по сравнению с инфантильной формой). Несколько позже присоединяются нарушения речи (смазанность, нечеткость), поперхивание при приеме пищи, а затем и при глотании воды. Голос приобретает гнусавый оттенок. Движения языком затрудняются. Все эти изменения формируют бульбарные и псевдобульбарные расстройства, а возникают в результате поражения ствола мозга. По утрам больных беспокоят неукротимые рвоты. Так же, как и при инфантильной форме, появляется мышечная слабость в конечностях, которая постепенно нарастает.
Мышечный тонус увеличивается, мышцы становятся плотными и твердыми на ощупь, появляются патологические стопные признаки (симптом Бабинского и другие). Постепенно эти изменения охватывают все четыре конечности, что становится причиной расстройств передвижения и самообслуживания. Возможны нарушение равновесия, расстройства поведения. Обычно интеллектуальные расстройства выражены незначительно или вообще отсутствуют, хотя описаны случаи и резкого снижения мыслительных способностей. У больных с ювенильной формой периодически регистрируют рефлекторную остановку дыхания: апноэ. В конце концов, прогрессирующее поражение нервной системы заканчивается смертельным исходом, в среднем, через 10 лет от появления начальных клинических признаков заболевания.
Взрослая форма развивается в сроки от 20 до 70 лет. Клинические симптомы довольно разнообразны, поскольку могут быть отражением патологии любого участка головного мозга. Чаще всего это парезы и параличи с повышенным мышечным тонусом, нарушения координации движений и равновесия, непроизвольные неконтролируемые движения, нарушения речи и глотания. Снижение интеллекта незначительное. Часто выявляется нистагм и нарушение содружественных (одновременных и однонаправленных) движений глазными яблоками. Болезнь прогрессирует и неизбежно заканчивается летальным исходом (обычно от присоединения интекуррентных инфекций).
Диагностика
Диагностика заболевания прижизненно довольно затруднительна, потому что клинических симптомов, свойственных только болезни Александера, нет. И специфических изменений ни один из методов исследования не выявляет (не считая генетического анализа, который, тем не менее, необходимо еще назначить, подозревая эту болезнь).
При магнитно-резонансной томографии головного мозга (МРТ) при болезни Александера выявляется демиелинизация различных отделов мозга (при инфантильной и юношеской формах — преимущественно в лобных с распространением на другие области, при взрослой — более выражена в мозжечке и стволе мозга).
При электроэнцефалографии регистрируют изменения биоэлектрической активности мозга в лобных отделах.
Генетический анализ наиболее точно позволяет подтвердить диагноз болезни Александера: находят мутацию в гене GFAP на 17-й хромосоме (в 95 % случаев). Следует помнить, что у 5 % больных этим заболеванием генетический дефект не обнаружен и по сей день.
Подтверждением заболевания служит обнаружение волокон Розенталя (что возможно при биопсии мозга или уже после смерти при вскрытии).
Лечение
Сегодня медицина не располагает эффективными способами лечения болезни Александера. Возможно, будущее в этом направлении принадлежит генной инженерии.
После установления диагноза обычно проводят симптоматическую терапию, позволяющую облегчить и продлить жизнь больному:
- при парезах назначают стимуляторы нервно-мышечной проводимости (Нейромидин);
- при спастичности мышц — миорелаксанты (Баклофен, Сирдалуд, Мидокалм);
- при эпилептических припадках — противосудорожные препараты (Вальпроаты, Сибазон и другие);
- для уменьшения непроизвольных движений могут использоваться нейролептики (Галоперидол, Азалептин и другие).
Для передвижения используют специальные приспособления, в том числе и ортопедические. Пик болезни позволяет передвигаться только с помощью инвалидной коляски. Конечно, в терминальных стадиях заболевания больные нуждаются в постоянном постороннем уходе.
Болезнь Александера — редкое, в основном, генетически обусловленное заболевание. Его развернутая клиническая картина представляет собой двигательные, координаторные нарушения, проблемы с речью и приемом пищи. Почти все взрослые больные живут не более 10 лет с момента развития заболевания. Наиболее точным методом диагностики является генетический. Способы лечения находятся в стадии разработки, больным в настоящее время помогают только симптоматическими средствами.[1]
Примечания
![]()
Эта страница в последний раз была отредактирована 11 февраля 2020 в 16:24.
Источник
Связанные заболевания и их лечение
Описания заболеваний
Национальные рекомендации по лечению
Стандарты мед. помощи
Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Классификация
- Диагностика
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Лейкодистрофия.
Лейкодистрофия
Описание
Лейкодистрофия. Нейродегенеративное заболевание, обусловленное наследственным нарушением обмена веществ с накоплением в головном и спинном мозге метаболитов, провоцирующих разрушение миелина. Манифестирует в основном в детском возрасте задержкой психомоторного развития, двигательными расстройствами, поражением зрительных и слуховых нервов, гидроцефалией, эпилептическими приступами. Диагностируется лейкодистрофия по данным неврологического статуса, анамнеза, генетических исследований, МРТ или КТ картины головного мозга, биохимических анализов. Лечение симптоматическое. При раннем выявлении и медленном прогрессировании возможна трансплантация пуповинной крови или костного мозга.
Дополнительные факты
Лейкодистрофия получила свое название в связи с поражением белого вещества мозга (с греческого leukos — белый). Различают около 60 разновидностей лейкодистрофии, определяющихся видом генной аномалии и возрастом манифестации клинических проявлений. Наряду с отдельными воспалительными поражениями ЦНС (например, лейкоэнцефалитом Шильдера) лейкодистрофия относится к синдрому диффузного склероза мозга. При этом доминирующее поражение миелина сближает ее с демиелинизирующими заболеваниями (рассеянным склерозом, РЭМ и пр. ), а отдельные формы можно отнести к липидозам.
К основным формам лейкодистрофии относятся метахроматическая, суданофильная, глобоидно-клеточная, дегенерация Ван-Богарта-Бертрана, болезнь Александера, вариант Галлервордена-Шпатца. Наиболее распространены первые 3 вида лейкодистрофии. Их встречаемость колеблется от 0,4 до 1 случая на 100 тыс. Новорожденных. Ряд форм лейкодистрофии являются настолько редкими, что в мировой литературе по неврологии описано всего несколько сотен их клинических наблюдений. В зависимости от возрастного периода, в котором дебютирует лейкодистрофия, каждая ее форма может подразделяться на инфантильный, поздний инфантильный, ювенильный и взрослый вариант.
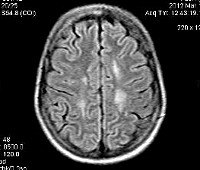
Лейкодистрофия
Причины
В своей основе каждая лейкодистрофия имеет генетическую аномалию определенного фермента. Вид аномалии и локализация генной мутации пока установлены лишь для наиболее встречающихся форм патологии. В большинстве случаев лейкодистрофия имеет аутосомно-рецессивный путь наследственной передачи, однако отдельные ее формы могут наследоваться сцеплено с полом. Кроме того, не одиноки случаи спонтанных мутаций. Генетически детерминированный энзимный дефект ведет к обменным нарушениям (чаще в метаболизме липидов) с отложением определенного метаболита в нервных структурах и отдельных соматических органах, в первую очередь в печени и почках.
Следствием метаболической аномалии является разрушение миелина оболочек нервных стволов и проводящих путей, гибель нейронов с замещением их разрастающейся глиальной тканью. Морфологически лейкодистрофия характеризуется диффузными и симметрично расположенными в полушариях головного мозга зонами гибели миелина, скоплением продуктов миелинового распада, усиленной пролиферацией глии. В отдельных нозологических вариантах лейкодистрофия имеет специфическую морфологическую картину — метахроматическое или суданофильное окрашивание продуктов миелинового распада, скопление в зонах демиелинизации глобоидных клеток.
Симптомы
В большинстве случаев лейкодистрофия дебютирует в раннем детском возрасте. Новорожденные, как правило, выглядят здоровыми. Определенный период они нормально развиваются, а затем постепенно возникают различные неврологические симптомы, отличающиеся неуклонным прогрессированием. Скорость нарастания симптомов тем выше, чем раньше манифестировала лейкодистрофия. Ведущими проявлениями выступают прогрессирующая олигофрения, ухудшение зрения, тугоухость, эписиндром, спастические парезы. Первыми симптомами лейкодистрофии могут быть атаксия, мышечно-тонические расстройства (гипо- или гипертонус, мышечные подергивания), экстрапирамидные проявления, изменения поведения. Затем возникают эпиприступы, бульбарные проявления, снижается слух и зрение, отмечается интеллектуальное снижение с постепенной утратой ранее приобретенных навыков. Сенсорные расстройства не характерны. На поздних этапах развития болезни наблюдаются параличи, выраженная олигофрения, грубое расстройство глотания, амавроз, глухота. В терминальной фазе обычно отмечается децеребрационная ригидность.
Слабость мышц (парез). Судороги. Тонико-клонические судороги. Тремор.
Классификация
Метахроматическая лейкодистрофия в зависимости от манифестации имеет 4 варианта. Врожденный вариант дебютирует в первые 1-3 мес. Жизни задержкой развития и судорожным синдромом; дети не достигают возраста 1 года. Позднедетский вариант метахроматической лейкодистрофии начинается в период от 1 до 3 лет с мышечной гипотонии и слабости, атаксии, задержки психического развития (ЗПР). Затем формируется спастическая тетраплегия, афазия, псевдобульбарный синдром. В редких случаях пациенты доживают до 10-летнего возраста. Ювенильный вариант манифестирует в 4-6 лет и длится в среднем 7 лет. Взрослый вариант дебютирует в третьей декаде жизни, иногда позднее, продолжительность жизни пациентов от начала клиники варьирует в пределах 10-20 лет.
Суданофильная лейкодистрофия наследуется сцеплено с Х-хромосомой и имеет несколько разновидностей. Лейкодистрофия Пелицеуса-Мерцбахера может стартовать на 1-ом году жизни или в 3-4 года. Первым признаком является крупноразмашистый нистагм, позже возникает ЗПР, мозжечковая атаксия, гиперкинезы, парезы. Наибольшее прогрессирование происходит в возрасте до 10 лет, затем заболевание принимает замедленное течение с длительными ремиссиями. Пациенты могут жить до зрелого возраста. Адренолейкодистрофия — вариант, при котором лейкодистрофия сочетается с надпочечниковой недостаточностью. Характеризуется прогрессирующим течением с летальным исходом спустя 6-8 лет от начала клиники.
Глобоидно. Клеточная лейкодистрофия (болезнь Краббе) — липоидоз с накоплением в очагах демиелинизации галактоцереброзида и образованием больших округлых глобоидных клеток. Раннедетский вариант развивается в первом полугодии жизни с гипервозбудимости и периодической гипертермии, задерживается психомоторное развитие, нарастает тонус мышц, затем развивается спастический тетрапарез, олигофрения, эписиндром, возможен опистотонус. В годовалом возрасте наступает летальный исход. Позднедетский вариант более редкий, манифестирует ухудшением зрения.
Спонгиозная дегенерация Ван. Богарта — Бертрана характеризуется эписиндромом, гиперсомнией, выраженной гидроцефалией с увеличением размеров головы, вызывающей амавроз атрофией зрительных нервов. Резкая внутричерепная гипертензия приводит к расхождению черепных швов, регистрируемому при рентгенографии черепа. Пациенты с этой формой лейкодистрофии погибают до 3-летнего возраста.
Болезнь Александера (лейкодистрофия с волокнистой формацией) обусловлена мутацией гена, ответственного за синтез GFAP белка. В результате происходит накопление в клетках глии аномального GFAP белка, содержащего волокна Розенталя. Неонатальный вариант имеет тяжелое течение с летальным исходом к концу 1-го года. Инфантильный вариант встречается примерно в половине случаев, проявляется в первые 1-2 года жизни ЗПР, затем присоединяются спастические парезы, атаксия, гидроцефалия. Дети погибают спустя несколько лет. Ювенильная лейкодистрофия Александера дебютирует в период от 4-х до 10-летнего возраста, протекает с преимущественно стволовой симптоматикой. Продолжительность жизни колеблется в пределах 10-30 лет. Взрослый вариант отличается поздней манифестацией и относительно медленным течением в пределах 10 и более лет.
Диагностика
Диагностический поиск требует привлечения ряда специалистов: невролога, педиатра, медицинского генетика, для диагностики расстройств зрения и слуха — отоларинголога и офтальмолога. Важное значение имеет изучение анамнеза болезни (возраст и симптомы дебюта, последовательность развития клиники) и семейного анамнеза (наличие лейкодистрофии у родственников). Нейросонография через родничок и эхо-энцефалография у пациентов более старшего возраста, как правило, выявляет повышение интракраниального давления. Лейкодистрофия сопровождается существенным увеличением концентрации белка, обусловленным разрушением церебральных клеток, что определяется при исследовании цереброспинальной жидкости.
С целью диагностики вида метаболической аномалии проводится целый ряд биохимических тестов с определением уровня ферментов и накапливающихся метаболитов. Очаги демиелинизации хорошо визуализируются при помощи МРТ, могут быть обнаружены и на КТ головного мозга. Обычно демиелинизация видна на МРТ головного мозга еще до клинической манифестации лейкодистрофии. Благодаря развитию генетики, лейкодистрофия имеет разработанную ДНК-диагностику, а отдельные ее формы (метахроматическая, адренолейкодистрофия, глобоидно-клеточная) — возможность пренатального диагностирования.
Лечение
На сегодняшний день лейкодистрофия не имеет эффективных способов терапии, позволяющих купировать прогрессирование симптомов. Проводится симптоматическое лечение — в основном дегидратационная и антиконвульсантная терапия. Единственным методом, способным увеличить продолжительность жизни пациентов с лейкодистрофией и улучшить качество их жизни, является трансплантация пуповинной крови или пересадка костного мозга. Трансплантация приводит к нормализации метаболизма. Однако этот процесс занимает длительное время (от 12 до 24 мес. ), в течение которого продолжается прогрессирование лейкодистрофии. Поэтому зачастую тяжелая инвалидизация или гибель пациента наступает даже после успешной трансплантации.
Следует подчеркнуть, что трансплантация никак не влияет на уже развившийся неврологический дефицит, она лишь позволяет приостановить его дальнейшее прогрессирование. В связи с тем, что эффект такого лечения наступает спустя 1-2 года, оно целесообразно в случае ранней доклинической диагностики лейкодистрофии (при соответствующей настороженности родителей рожденного ребенка в связи с наличием подобной патологии в семье) или при медленно прогрессирующем варианте течения. Кроме того, необходимо учитывать, что трансплантация связана с риском ряда серьезных осложнений, таких как отторжение, реакция «трансплантат против хозяина», развитие инфекций.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник