Бета талассемия код по мкб 10

Рубрика МКБ-10: D56.1
МКБ-10 / D50-D89 КЛАСС III Болезни крови, кроветворных органов и отдельные нарушения, вовлекающие иммунный механизм / D55-D59 Гемолитические анемии / D56 Талассемия
Определение и общие сведения ( в т.ч. эпидемиология)[править]
Талассемии — наследственные заболевания, характеризующиеся нарушением синтеза альфа- или бета-цепей гемоглобина.
Бета-талассемия
Синонимы: анемия Кули, средиземноморская анемия
Бета-талассемия характеризуется дефицитом (Beta+) или отсутствием (Beta0) синтеза бета-глобиновых цепей гемоглобина (Hb).
Бета-талассемия чаще всего встречается у выходцев из Средиземноморья, Среднего Востока и Индии, а альфа-талассемия— у выходцев из Африки, Юго-Восточной Азии, Средиземноморья и Среднего Востока.
Ежегодная заболеваемость симптоматическими формами бета-талассемии оценивается в 1/100 000 во всем мире и 1/10 000 в Европе. Распространенность не известна.
Наследование является аутосомно-рецессивным.
Этиология и патогенез[править]
Бета-талассемия вызвана точечными мутациями или, реже, делециями в гене HBB (11p15.5), что приводит к уменьшению (бета+) или отсутствию (бета0) синтеза бета-цепей гемоглобина. Мутации, вызывающие большую бета-талассемию, являются гомозиготными или компаундными гетерозиготными.
Клинические проявления[править]
Выделяют 3 основных формы бета-талассемии:
Малая бета-талассемия (гетерозиготная форма) проявляется легкой микроцитарной гипохромной анемией. Анамнез не имеет диагностического значения, за исключением типичной этнической принадлежности и наличия микроцитарной анемии у кого-либо из родителей. Физикальное исследование тоже малоинформативно. Диагностическим признаком служит повышение гемоглобина A2. Если талассемия сочетается с дефицитом железа, уровень гемоглобина A2 снижен, что затрудняет диагностику. Примечательно, что у больного родителя также имеется микроцитоз и повышение уровня гемоглобина A2. Хотя малая бета-талассемия не требует лечения, показана генетическая консультация в связи с возможностью гомозиготного заболевания у других детей.
Большая бета-талассемия (гомозиготная форма) протекает тяжело вследствие гемолиза и нарушенного эритропоэза. Хотя тяжесть анемии варьирует, в большинстве случаев жизненно необходимо переливание эритроцитарной массы. Угрожающая жизни большая бета-талассемия известна как анемия Кули.
У некоторых гомозигот наблюдается промежуточная бета-талассемия с менее выраженной симптоматикой.
Бета-талассемия: Диагностика[править]
Анамнез и физикальное исследование
При сборе анамнеза часто выявляют типичную этническую принадлежность и случаи микроцитарной анемии в семье. В первые месяцы жизни большая бета-талассемия клинически не проявляется в связи с наличием фетального гемоглобина (HbF). Симптомы появляются обычно в возрасте 6—9 мес, а при повышенном уровне фетального гемоглобина или менее тяжелом течении — несколько позже. Характерны прогрессирующая анемия, задержка физического развития, гепатоспленомегалия, нарушение роста костей вследствие гиперплазии костного мозга (наиболее заметны макроцефалия и деформация лицевого черепа).
Лабораторные и инструментальные исследования
Обнаруживают признаки тяжелой микроцитарной гипохромной анемии, в мазке периферической крови — выраженный ретикулоцитоз, полихромазию, эритрокариоциты, мишеневидные эритроциты и базофильную зернистость эритроцитов. При электрофорезе выявляют снижение или отсутствие гемоглобина A1, повышение уровня гемоглобина A2 и, нередко, — фетального гемоглобина. Уровень сывороточного железа часто повышен в связи с усиленным всасыванием его в ЖКТ. На рентгенограммах костей видны гиперплазия костного мозга и истончение компактного вещества, особенно костей черепа. У обоих родителей имеется микроцитарная анемия и повышение уровня гемоглобина A2.
Дифференциальный диагноз[править]
Дифференциальный диагноз обычно несложен и может включать генетические сидеробластические анемии, врожденные дизэритропоэтические анемии и другие состояния с высоким уровнем HbF (такие как ювенильный миеломоноцитарный лейкоз и апластические анемии).
Бета-талассемия: Лечение[править]
Без лечения большая бета-талассемия с тяжелым течением всегда приводит к смерти. Постоянные переливания эритроцитарной массы продлевают жизнь, однако в конце концов они вызывают гемосидероз печени, эндокринных желез, сердца. Без терапии комплексобразующими средствами смерть наступает в возрасте 20—30 лет из-за гемосидероза.
а) Переливание эритроцитарной массы, обедненной лейкоцитами, 10—20 мл/кг, проводят каждые 3—5 нед, чтобы гематокрит не опускался ниже 30%. Этого достаточно для нормального роста и развития костей.
б) Дефероксамин назначают по достижении полного насыщения трансферрина, когда пробное введение препарата выявляет несвязанное железо (см. гл. 4, п. II.В.3.в.1). Как правило, трансферрин полностью насыщается, когда общий объем перелитой эритроцитарной массы превышает 500 мл/кг. Доза дефероксамина — 30—50 мг/кг/сут п/к в течение 10—12 ч ночью с помощью инфузионного насоса. Дозу корректируют в зависимости от избытка железа, скорости его экскреции и количества железа, связанного дефероксамином. Такое лечение значительно уменьшает риск гемосидероза внутренних органов и продлевает жизнь. Препараты железа противопоказаны.
в) Спленэктомия выполняется при спленомегалии для уменьшения потребности в переливаниях крови. По возможности операцию откладывают до 5-летнего возраста, чтобы уменьшить риск сепсиса. Предварительно проводят вакцинацию против пневмококков, Haemophilus influenzae типа B и менингококков, а после операции профилактически назначают антибиотики (см. табл. 14.2). В случае лихорадки требуется тщательное обследование для предотвращения и лечения сепсиса.
г) Фолиевую кислоту , 1 мг/сут, назначают тем, кому не проводят постоянные переливания крови, например при промежуточной талассемии. Это помогает предотвратить дефицит фолиевой кислоты и мегалобластный криз, которые вызываются усиленным эритропоэзом.
д) Трансплантация костного мозга может привести к излечению, но из-за осложнений и высокой смертности эту операцию проводят лишь в немногих центрах. Костный мозг берут у доноров-родственников, совместимых по HLA.
Профилактика[править]
Чтобы предотвратить рождение детей с тяжелыми альфа- и бета-талассемиями, проводят генетическое консультирование и пренатальную диагностику.
Прочее[править]
Прогноз
Прогноз зависит от тяжести состояния, но, как правило, хороший, особенно при условии адекватного лечения.
Источники (ссылки)[править]
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
- Луспатерцепт
Источник
Содержание
- Описание
- Симптомы
- Причины
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
Талассемия.
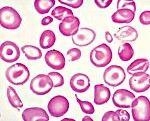
Талассемия
Описание
Талассемии представляют собой гетерогенную группу гемоглобинопатии, в основе которых лежит снижение синтеза полипептидных цепей, входящих в структуру нормального гемоглобина А. Талассемия — это мишеневидноклеточная анемия с нарушенным соотношением НЬА и HbF по биохимическим показателям; при этом возможна частичная недостаточность определенной цепи или ее полное отсутствие при преобладании другой цепи. Так, при нарушении синтеза ß-цепи будут преобладать а-цепи и наоборот. Бета-талассемия обусловлена снижением продукции ß-цепей гемоглобина. Неповрежденные а-цепи избыточно накапливаются в клетках эритропоэза, что ведет к повреждению мембраны и разрушению как клеток эритроидного ряда в костном мозге, так и эритроцитов в периферической крови; развиваются неэффективный эритропоэз и гемолиз с гипохромией эритроцитов, ибо содержание гемоглобина в эритроцитах недостаточно. Первыми описали ß-талассемию американские педиатры Кули и Ли в 1925 г. Тяжелая гомозиготная форма ß-талассемии получила название болезни Кули, или большой талассемии. Кроме того, по выраженности анемии и других клинических симптомов выделяют промежуточную, малую и минимальную талассемию. Помимо стран Средиземноморья талассемия встречается во Франции, Югославии, Швейцарии, Англии, Польше, а также у жителей Закавказья и Средней Азии, где в некоторых регионах частота носительства достигает 10-27 %.
Симптомы
Клиника большой талассемии проявляется уже в детстве. У больных детей своеобразный башенный череп, монголоидное лицо с увеличенной верхней челюстью. Ранний признак болезни Кули — сплено- и гепатомегалия, развивающиеся за счет экстрамедуллярного кроветворения и гемосидероза. Со временем у них формируются цирроз печени, сахарный диабет в результате фиброза поджелудочной железы, а гемосидероз миокарда приводит к застойной сердечной недостаточности. Гомозиготная бета-талассемия(большая талассемия, анемия Кули) характеризуется резким снижением образования HbA1, значительным увеличением содержания HbF, низким, нормальным или повышенным содержанием НbA2. Содержание НbF может колебаться от 30 до 90 %, иногда бывает ниже 10%. Течение заболевания характеризуется тяжелой гемолитической анемией, проявляющейся к концу первого года жизни ребенка, гепато- и спленомегалией, монголоидиостью лица и башенным черепом, отставанием ребенка в физическом развитии, нередко желтушностью и бледностью кожных покровов. У части больных развиваются язвы в области голеней. Рентгенологически обнаруживают симптом «ежика» или «щетки», который положителен при увеличении содержания HbF, отрицателен при увеличении процента НbA2. У детей в возрасте от 6 мес. До 1 года в мелких костях стоп и кистей выявляется истончение коркового слоя со вздутием кости и образованием грубосетчатой структуры костного мозга. Начиная с 1-го года жизни ребенка отмечается нарушение развития костей, быстро прогрессирующее до периода полового созревания. Длительно продолжающийся гемолиз (ретикулоцитоз, увеличение свободной фракции билирубина сыворотки крови, уробилинурия, гиперсидеремия), частые переливания эритроцитной массы приводят к развитию гемосидероза печени и селезенки. Нередко происходит образование билирубиновых камней в желчных путях. Уровень гемоглобина достигает 30-50 г/л, цветовой показатель 0>5 и ниже. В мазках крови обнаруживают мишеневидные эритроциты, отличающиеся малым содержанием гемоглобина и укорочением продолжительности жизни, анизопойкилоцитоз, эритро- и нормобласты. Отмечается повышение осмотической стойкости эритроцитов, лейкопения (в период гемолитического криза). В костном мозге — раздражение эритро-нормобластического ростка. Иногда возникают апластический криз или явления гиперспленизма. При тяжелой гомозиготной талассемии больные умирают на первом году жизни, при сравнительно более спокойной форме заболевания они могут дожить до взрослого возраста. Гетерозиготная бета-талассемия протекает в виде как бессимптомной, так и манифестной форм с незначительно увеличенной селезенкой, специфическими костными изменениями, нередко выраженной гипохромной анемией, часто анизоцитозом, пойкилоцитозом и мишеневидностью эритроцитов, повышенной их осмотической резистентностью увеличением количества НbA2 (примерно до 8% от общего гемоглобина), у части больных — HbF (до 5%). При гетерозиготной дельтабета-талассемии (F) отмечается высокое содержание HbF при нормальном уровне НbA2. Клинические признаки и гематологические сдвиги аналогичны встречающимся при гетерозиготной бета-талассемии. Гомозиготные формы дельтабета-талассемии (F) проявляются почти теми же клинико-гематологическими нарушениями, что и гомозиготная бета-талассемия. У больных с этой формой заболевания обнаруживается только HbF. Среди больных талассемией удается выделить лиц с гетеро-и гомозиготными формами А2F-талассемии, которые по признакам, характеризующим их течение, по существу мало отличаются от бета-талассемии. В группе больных бета-талассемией случаи большой талассемии с выраженными клиническими проявлениями встречаются реже, чем промежуточные и малые формы. При обследовании родственников больных чаще обнаруживается минимальная форма бета-талассемии. Выделяют следующие формы а-талассемии: водянка плода с гемоглобином Bart’s (у4). Гемоглобинопатия Н (бета4), а-талассемия-1 и а-талассемия-2. Водянка плода представляет собой гомозиготное состояние (по генам а-th-l), несовместимое с жизнью. Беременность в подобных случаях непроизвольно прерывается и у плода выявляют водянку мозга, гепатомегалию. Электрофоретическим исследованием гемоглобина обнаруживается Hb Bart’s (80-90%, сочетающийся со следами НbН. Гемоглобинопатия Н — один из вариантов а-талассемии — проявляется гемолитической анемией, увеличением селезенки, тяжелым течением костных изменений. Картина периферической крови характеризуется понижением содержания гемоглобина, анизо- и пойкилоцитозом, гипохромией и множественными включениями в эритроцитах (выпавший в осадок гемоглобин Н). Гетерозиготные формы а-талассемии выявляются у родственников больных гемоглобинопатией Н. А-Талассемия-1 (малая форма заболевания) возникает при сочетании гена а-th-l с нормальным геном a-цепочкового синтеза. Она характеризуется небольшой анемией, умеренным анизо- и пойкилоцитозом, внутриэритроцитарными включениями, повышенной осмотической резистентностью эритроцитов. У взрослых больных а-талассемией-1 гемоглобиновые фракции бывают в пределах нормы, у новорожденных выявляется Hb Bart’s (5-10%). А-Талассемия-2 (минимальная форма заболевания) развивается при сочетании гена a-th-2 с нормальным геном а-цепочкового синтеза. Клинические проявления отсутствуют.
Гипербилирубинемия. Ломота в мышцах. Низкая температура тела. Ретикулоцитоз.
Талассемия
Причины
При талассемии нарушается синтез одной из четырёх цепей глобина. Наследование патологии от одного (гетерозиготность) или обоих родителей (гомозиготность), тип нарушенной цепи определяют выраженность клинических проявлений. Причины повышенной гибели эритроцитов связаны с нарушенной структурой клетки из-за неправильного соотношения цепей глобина в гемоглобине. Кроме укорочения жизни эритроцитов при данном заболевании происходит гибель клеток предшественников эритроцитов в костном мозге.
Лечение
Переливания эритроцитов. При тяжелых формах талассемии потребность в переливаниях эритроцитарных препаратов крови возникает уже с первых месяцев жизни и сохраняется, хотя и в разной степени, пожизненно – развивается так называемая трансфузионная зависимость. Это означает, что гемоглобин в крови больных постоянно продолжает снижаться и других реальных способов его повышения, кроме таких переливаний, нет. Желательно, чтобы в крови больного содержание гемоглобина не падало до низких цифр, лучше осуществлять повторное переливание при еще удовлетворительных его уровнях- 95-100г/л. Дело в том, что при выраженном снижении гемоглобина активизируются многие присущие именно талассемии патологические процессы: например, упоминавшееся избыточное патологическое костеобразование, увеличение размеров печени и селезенки; ухудшается функция всех органов, снижается сопротивляемость к инфекциям из-за усилившегося кислородного голодания. При большой b-талассемии кроме замещения недостатка эритроцитов в циркулирующем кровяном русле с помощью переливаний эритроцитарных компонентов крови достигаются подавление собственного избыточного, но малоэффективного кровообразования в костном мозге больного, а также уменьшается всасывание железа в кишечнике. Таким образом, в наблюдении за больным талассемией важным является недопущение развития эпизодов выраженного падения уровня гемоглобина – это, во-первых, непосредственно может угрожать жизни, а во-вторых, способствует прогрессированию патологических проявлений талассемии. В то же время переливание эритроцитарных препаратов крови имеет свои существенные минусы. Как известно, при переливаниях препаратов крови обязательно учитывается совместимость донора и реципиента по группе крови, резус фактору. Но поскольку не существует в природе генетически одинаковых людей, то при повторных переливаниях организм пациента раньше или позже начинает вырабатывать белки-антитела, реагирующие с другими, более сложными частями мембран переливаемых кровяных телец (эритроцитов). Поэтому через некоторое время (обычно 3-4 года) организм больного становится биологически «совместимым» уже не с любым донором, подходящим по группе крови и резус-фактору, а только с определенными донорами, обладающими специфическим набором белков-антигенов на эритроцитах. Поэтому переливания эритроцитарных сред при талассемии желательно проводить по индивидуальному подбору, осуществляемому специальной изосерологической лабораторией станций переливания крови. Кроме того, эритроцитарные среды для больных талассемией должны быть специально очищенными от других биологических компонентов, содержащихся в крови (лейкоциты, многочисленные белки плазмы), поскольку они являются причиной т. Н. «пирогенных» реакций, проявляющихся ознобами и повышениями температуры, нередко до высоких цифр. Цельную кровь в настоящее время не переливают, переливания необработанной дополнительными методами эритроцитной массы также не желательны. В качестве высокоочищенных эритроцитарных сред сейчас используются размороженные, отмытые или фильтрованные эритроциты, которые гораздо реже вызывают реакции. Но как бы то ни было, при наличии трансфузионной зависимости переливания неизбежны, поэтому можно лишь принимать меры к уменьшению их побочных действий и предупреждению реакций и осложнений. При более легких формах талассемии, когда у больных либо имеется анемия легкой степени (на уровне 90-110г/л), либо гемоглобин в норме, а главное – что он стабильно сохраняется у больного с течением времени и не продолжает неуклонно снижаться, переливания препаратов крови не проводятся. Десферал. Важной частью лечения является выведение избытка железа из организма с помощью препаратов из группы «хелатов» (т. Н. «хелатная терапия»), осуществляемая препаратом «десферал». В настоящее время принято лечение подкожными многочасовыми инъекциями, наиболее удобно применение специальных аппаратов — т. Н. Помп, которые прикрепляются к одежде. Из фиксированного в помпе шприца десферал за несколько часов постепенно вводится подкожно пациенту. В идеале больные тяжелой формой талассемии должны получать десферал на протяжении всей жизни по 5 дней в неделю, но в реальной жизни это пока труднодостижимо. В странах с большой распространенностью талассемии, особенно высокоразвитых (например, в Италии) существуют специальные государственные программы помощи больным талассемией, которые предусматривают, кроме другого лечения, обеспечение необходимым для них десфералом и помпами для его введений. Из стран бывшего СССР аналогичная программа существует в Азербайджане. Хранить десферал следует в темном месте при +8-15˚С, разводить непосредственно перед началом введения. С осторожностью назначается лечение десфералом у детей младше 2–х лет ввиду относительно большего риска развития побочных эффектов лечения. У таких детей лечение десфералом начинают, если уже проведено около 15-20 переливаний препаратов крови, т. Е. Потребность в переливаниях уже довольно большая. Для улучшения качества жизни больных предпочтительна инфузия десферала ночью. Следует систематически менять места подкожных инъекций во избежания местного повреждения кожи и подлежащих мягких тканей. Как и любое лечение, терапия десфералом имеет свои возможные побочные эффекты. Чаще встречаются аллергические реакции на препарат, возможна также лихорадочная реакция. В случае появления на фоне лечения десфералом каких-либо новых жалоб следует обратиться к врачу, он решит вопрос о продолжении лечения и о том, каким образом лечить побочные эффекты десферала. Удаление селезенки (спленэктомия) У некоторых больных очень большие размеры селезенки сами по себе начинают негативно сказываться на состоянии гемоглобина и других показателей системы крови. В таких случаях проводят ее хирургическое удаление. От самой талассемии эта операция не излечивает, хотя и может смягчить ее проявления (что, впрочем, может и не произойти). Спленэктомия проводится только при очень больших размерах селезенки, а также когда имеют место явные признаки ее патологического действия на другие показатели крови (так называемый «гиперспленизм»), Операция не целесообразна ранее достижения возраста 5 лет, оптимальным считается возраст 8-10 лет. Первый год обычно наблюдается хороший эффект, но затем возможно рецидивирование проявлений талассемии, может нарасти увеличение печени. Кроме того, возрастает инфекционный риск, особенно относительно присоединения т. Н. «пневмококковой» инфекции в виде сепсиса, пневмонии. В связи с этим обязательна вакцинация против пневмококка, проводимая предпочтительно в предоперационный период. Вообще же решение вопроса об удалении селезенки всегда должно приниматься с большой осторожностью. Пересадка (трансплантация) костного мозга В настоящее время все большее распространение получает лечение талассемии с помощью пересадки костного мозга. Это единственный метод радикального лечения талассемии. При выявлении талассемии желательно, чтобы пациенты и члены их семей были «типированы по системе HLA» (т. Е. Прошли довольно сложное биологическое обследование на совместимость) с целью поиска возможного донора костного мозга. Однако найти подходящего донора обычно сложно, сама процедура поиска совместимого неродственного донора пока остается дорогой и длительной по времени. Очень дорога и сама пересадка костного мозга. Близкие родственники, даже если являются совместимыми по антигенам HLA, сами нередко имеют талассемию. Поэтому реальными кандидатами на лечение пересадкой костного мозга пока еще становятся относительно немногие больные талассемией. Следует отметить, что результаты пересадки костного мозга во многом зависят от качества проводившейся больному ранее терапии. Лучше результаты пересадки костного мозга у детей. Хотя разработка и внедрение новых методов лечения талассемий, включая пересадку костного мозга, продолжается, все же для подавляющего большинства больных реально возможными пока остаются приводившиеся выше «традиционные» методы лечения. В настоящее время также разрабатываются методы лечения талассемии с помощью генной инженерии. Больным талассемией следует соблюдать диету (стол №5). Полезны напитки, содержащие танин: чай, какао, а также орехи, соя – эти продукты уменьшают всасывание железа. Из-за склонности к кариесу рекомендуются фтористые зубные пасты, своевременная санация полости рта. Для улучшения функции печени врач назначает лекарства – т. Н. «гепатопротекторы». К ним относится липоевая кислота, вит Е, препараты типа хорошо известного «эссенциале». Улучшает выведение железа из организма аскорбиновая кислота (витамин С) в дозе 50мг/сутки до 10 лет и 100мг/сутки у детей старше 10 лет. Также применяется лечение витаминами группы В, фолиевой кислотой. Увеличение дозы витаминов проводят при стрессе, беременности. Назначаются курсы желчегонных трав — мята, овес, кукурузные рыльца, барбарис, а также тюбажи. Особенности лечения других форм талассемии «Промежуточная» талассемия. Вследствие более мягкого течения заболевание не требует постоянных переливаний — обычно не чаще 1 раза в 2-3нед – 2-3мес. При присоединении интеркуррентных заболеваний, при операциях- проводятся переливания эритроцитарных сред при уровне гемоглобина ниже 70 г/л. Как правило, назначается лечение десфералом, но предпочтительнее после специального исследования обмена железа по уровню его содержания в крови и ответу на разовое введение десферала (т. Н. «десфераловый тест»). При больших размерах селезенки с признаками избыточно повышенной функции рассматривается вопрос об ее оперативном удалении «Малая» талассемия. Переливаний эритроцитов не требует. При анемии назначают фолиевую кислоту, также возможно решение вопроса о лечении десфералом по уровню сывороточного железа или десфераловому тесту. При тяжелых формах талассемии, особенно с наличием трансфузионной зависимости, больному оформляется инвалидность.
Основные медуслуги по стандартам лечения | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Клиники для лечения с лучшими ценами
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Источник