Аутовоспалительный синдром что это такое

 Дифференциальная диагностика при лихорадках неясного генеза всегда представляет значительные трудности для точного установления заболевания. Причинами такой лихорадки могут быть инфекционные, онкологические, аутоиммунные заболевания. Кроме того, в качестве возможных причин длительной лихорадки, особенно при рецидивирующем ее характере, рассматриваются так называемые периодические синдромы.
Дифференциальная диагностика при лихорадках неясного генеза всегда представляет значительные трудности для точного установления заболевания. Причинами такой лихорадки могут быть инфекционные, онкологические, аутоиммунные заболевания. Кроме того, в качестве возможных причин длительной лихорадки, особенно при рецидивирующем ее характере, рассматриваются так называемые периодические синдромы.
Основные причины
Изучение природы периодических синдромов лихорадки показало, что в их основе лежит гиперактивация неспецифического (врожденного) иммунитета, что приводит к развитию спонтанного системного воспаления, с лихорадкой и привлечением многих органов, но при этом отсутствуют аутоантитела или любые другие признаки аутоиммунитета. На основе этого наблюдения был предложен термин «аутовоспалительные заболевания / синдромы» (АС). Наличие у больных клиники периодической лихорадки дало предварительное название этой группе заболеваний – “синдромы периодической лихорадки” (СПЛ).
АС относятся к группе системных заболеваний. В настоящее время они являются предметом активного изучения во всем мире. Если сначала в эту группу были включены только патологии с генетическими дефектами, обусловившие дисрегуляцию врожденного иммунитета, то сейчас список дополнен идиопатическими, а также полигенными заболеваниями, которые имеют черты аутовоспаления. Единой классификации аутовоспалительных синдромов нет, список этих патологий постоянно обновляется.
1. Генетические синдромы СПЛ:
– средиземноморская семейная лихорадка (ССМЛ);
– циклический синдром TRAPS;
– гипер IgD-синдром;
2. Другие моногенные АС:
– криопирин-ассоциированный;
– семейный холодовой;
– Макла-Уэллса;
– синдром CINCA / NOMID;
– PAPA-синдром;
– ювенильный системный гранулематоз (синдром Блау);
– дефицит антагониста рецептора ИЛ-1;
– мевалоновая ацидурия;
– синдром Маджида;
– хронический циклический мультифокальный остеомиелит.
3. Ненаследственные или полигенные расстройства:
– синдром Шнитцлера;
– патология Бехчета;
– синдромы PFAPA и Маршалла;
– ювенильный ревматоидный артрит с системным началом;
– болезнь Стилла с началом во взрослом возрасте.
Виды заболеваний
Аутовоспалительные заболевания являются достаточно редкими заболеваниями, практически все они начинаются в детском возрасте.
Рассмотрим некоторые из них.
ССМЛ (пароксизмальный полисерозит, семейный рецидивирующий полисерозит, периодическая болезнь (ПБ)).
Это наследственная болезнь, которая встречается среди лиц, относящихся к средиземноморским этногруппам (евреи-сефарды, арабы, турки, армяне, итальянцы) и определяется короткими приступами лихорадки, вызванными индуцированными нейтрофилами и воспалением серозных оболочек с постепенным развитием амилоидоза почек.
Генетическая основа заболевания – мутации в гене MEFV, кодирующего синтез регуляторного белка Пирина. Пирин является противовоспалительными агентом.
Рецидивирующее воспаление, которым характеризуются нападения ПБ, связывают с дегрануляцией клеток, соответственно, действуют иммунологические механизмы аллергического или неаллергического типов. Вследствие уменьшения или отсутствия противовоспалительного эффекта Пирина в крови активизируется противовоспалительный цитокин – интерлейкин-1 (IL-1), что приводит к накоплению в крови воспалительных белков, в том числе С-реактивного белка, 1- глобулина. Последний белок и является предшественником АА-амилоида и приводит к развитию амилоидоза при ССМЛ.
Приступы лихорадки чаще проявляются в первые 20 лет жизни, реже – в пожилом возрасте. Частота и продолжительность приступов разная: у одних возникают один раз в 2 недели, у других могут быть один раз в год. У некоторых лиц нападки не повторяются годами. Как правило, они не наблюдаются в период беременности.
Острые приступы проявляются внезапным подъемом температуры тела – до 39-40 С. Температура обычно сочетается с признаками серозита. Чаще всего (почти в 95%) имеет место поражение брюшной полости с картиной перитонита. Часто это локальная боль, которая затем распространяется на всю брюшную полость. Одновременно может развиться Дефанс (напряжение) мышц живота с картиной заворота кишок.
У многих больных могут быть проявления плеврита и артрита. При длительных частых приступах могут отмечаться такие же изменения, как и при деформирующем остеоартрите. У 25% пациентов может проявиться узловатая эритема.
Диагностика
Диагностическими критериями предлагаются следующие.
Критерии предполагаемого диагноза:
– периодически обостренно возникающие фебрильные синдромы перитонита и / или плеврита (артрита) с наступлением ремиссии в срок до 1 нед. даже без всякого лечения;
– в определенной степени присутствует значимость этнонациональной принадлежности;
– достаточно часто встречаются признаки наследственного характера болезни;
– для женского пола – ремиссия при беременности и рецидивы после родов;
– позитивная проба с внутривенным введением метараминола (при введении этого препарата у больных возникает типичный приступ ПБ, чего не наблюдается у здоровых людей).
Верифицирующие критерии: выявление мутации гена MEFV. Проведение молекулярно-генетических исследований при отсутствии «больших» и наличии только дополнительных критериев ПБ (например, принадлежность к определенной этнической группе) или лабораторной и ультразвуковой симптоматики наиболее тяжелого осложнения данного заболевания – амилоидоза почек позволяет иногда впервые поставить диагноз. Порой встречаются пациенты с ПБ, которая протекает в виде лихорадки без тканевых поражений. В этом случае нередко устанавливают диагноз «лихорадка неясного генеза» У больных без амилоидоза прогноз для жизни благоприятный.
В развитии амилоидоза важную роль играет нарушенное торможение воспаления, поскольку при дефиците / дефекте растворимый рецептор TNF вовремя не блокируется, а воспаление становится неконтролируемым. В перечень симптомов входят: короткие повторные эпизоды фебрильной лихорадки, которые продолжаются в течение 4-6 дней и сочетаются с сильной болью в животе, тошнотой и рвотой; возникновение олигоартрита, миалгии (сыпь на коже), конъюнктивита и одностороннего периорбитального отека. Эпизоды болезни иногда продолжаются до 3 нед. и более. К провоцирующим факторам относятся физическая нагрузка, эмоциональный стресс. Амилоидоз развивается у 25% пациентов. При диагностике отмечается повышение показателей, характерных для заполнения в период приступов болезни, но специфическим признаком является снижение уровня растворимого рецептора TNF1А и повышение уровня TNF.
Синдром Макла-Уэллса
Возникает заболевание в возрасте до 5 лет и, как правило, завершается в подростковом возрасте. Это доброкачественная болезнь.
Системный ювенильный артрит
Существует тяжелая форма среди всех вариантов течения ювенильного идиопатического артрита (ЮИА) У больных, в частности, у детей с системным ЮИА (сЮИА) в активном периоде болезни выявлено существенное завышение уровня IL-6, IL-1 и IL-1Ra в сыворотке крови. Цитокиновая дисрегуляция во врожденном иммунитете с подъемом уровней IL-6 и IL-1Ra является ключевым механизмом патогенеза у больных сЮИА.
Заболевание диагностируют при наличии:
– персистирующей лихорадки 39 С и выше продолжительностью дольше двух недель;
– артрита одного и более суставов дольше 6 недель;
– высыпания;
– лимфаденопатии;
– перикардита;
– гепатоспленомегалии.
Типичным для сЮИА является наличие в лабораторных обследованиях нейтрофильного гиперлейкоцитоза, гипертромбоцитоза, а также положительных показателей острой фазы воспаления. Повышение уровня IL-6 и IL-1Ra часто используют для верификации диагноза сЮИА.
Итак, аутовоспалительные синдромы являются моногенными заболеваниями, объединяющими СПЛ.
Источник
, ,
АУТОВОСПАЛИТЕЛЬНЫЕ СИНДРОМЫ – «НОВАЯ» МУЛЬТИДИСЦИПЛИНАРНАЯ ПРОБЛЕМА ПЕДИАТРИИ И РЕВМАТОЛОГИИ.
ФГБУ «НИИР» РАМН (ФЕДЕРАЛЬНОЕ ГОСУДАРСТВЕННОЕ БЮДЖЕТНОЕ УЧРЕЖДЕНИЕ «НАУЧНО-ИССЛЕДОВАТЕЛЬСКИЙ ИНСТИТУТ РЕВМАТОЛОГИИ» РАМН), Москва
В последние годы в клинической практике и в различных публикациях нередко приходится сталкиваться с так называемыми аутовоспалительными заболеваниями, относящимися к редко встречающимся и являющихся сложными для диагностики и лечения. Аутовоспалительные заболевания/ синдромы человека (Human Autoinflammatory Disease) – АВС/HAIDS — гетерогенная группа редких генетически детерминированных, наследственно обусловленных состояний, характеризующихся периодическими приступами воспаления и манифестирующихся лихорадкой и клинической симптоматикой, имитирующей ревматическую при отсутствии аутоиммунных или инфекционных причин [P. Fietta 2004] [1].
Проблема АВС вызывает большой интерес у исследователей всего мира, она часто обсуждается на международных форумах, конференциях и уже нашла широкое освещение в литературе, преимущественно зарубежной [1,2,3,4]. На прошедшем в сентябре 2011 года в Бельгии XVIII Европейском педиатрическом конгрессе ревматологов также детально обсуждались вопросы, касающиеся АВС у детей.
Распознавание этих синдромов представляет большие диагностические трудности, с которыми может в реальной клинической практике столкнуться педиатр, ревматолог и врачи других специальностей, в силу чего они должны располагать сведениями об этой патологии.
Список болезней группы АВС за время их изучения постоянно расширялся и обновляется до настоящего времени. Имеется несколько вариантов классификации АВС, один из них представлен в табл. 1 [1].
Согласно проекту EUROFEVER, созданному группой исследователей в 2002 году в рамках педиатрического ревматологического европейского сообщества (PRеS)/PRINTO к числу этих заболеваний добавлены еще ряд состояний таких как CINCA/NOMID синдром, хронический рецидивирующий мультифокальный остеомиелит (CMRO), DIRA и другие (табл.2). Одним из важных результатов деятельности EUROFEVER проекта стало создание международного регистра пациентов с АВС, основной целью которого является накопление информации о пациентах, клинических проявлениях, исходах и терапии, обеспечение доступности информации для врачей и пациентов [5]. В целом к 2011 году в регистр были включены 1970 пациентов из 32 стран, в том числе 20 из России [6].
Установлено, что АВС чаще всего дебютируют в детском возрасте, иногда на первом году жизни и являются поэтому, главным образом, педиатрической проблемой. В то же время, эти заболевания могут начинаться и у взрослых, или, дебютируя у детей, сопровождать больного на протяжении многих лет жизни. Большинство синдромов из указанной группы (семейная средиземноморская лихорадка, синдром гипер — IgD, синдром PFAPA, синдром СINCA и др.) относятся к редкой патологии, в силу чего рассматривались нами ранее при изучении лихорадки неясного генеза (ЛНГ) как редкие ревматические заболевания [7]. Эта «редкость» во многом обусловлена трудностью диагностики и недостаточным знанием указанных заболеваний педиатрами. Между тем как реальный шанс встретить пациента с подобной патологией есть у каждого врача.
Ведущий признак АВС — рецидивирующий лихорадочный синдром, являющийся интригующей и загадочной проблемой, волнующей умы исследователей не одно поколение. Именно он представляет сложную дифференциально-диагностическую задачу даже для опытного клинициста и нередко для своего решения требует мультидисциплинарного подхода с привлечением специалистов различного профиля. Для верификации диагноза используется разработанный нами ранее для ЛНГ диагностический алгоритм, предусматривающий исключение широкого спектра инфекционных (бактериальных и вирусных) заболеваний, сопровождающихся периодическим лихорадочным синдромом, а также гематоонкологических, ревматических, аутовоспалительных и других патологических состояний.
Общими клинико-лабораторными проявлениями АВС являются также мышечно-артикулярная симптоматика, разнообразный характер сыпи, воспаление серозных оболочек, высокие лабораторные показатели активности воспаления (СОЭ, лейкоциты, фибриноген, SAA), возможное развитие амилоидоза, отсутствие аутоантител или активации аутоспецифических клеток. Причина (для большинства синдромов) — наличие одного мутантного гена, наличие генов – «модификаторов» течения заболевания, мутации которых меняют течение заболевания (утяжеляют или ослабляют). Основное звено патогенеза – гиперактивация естественного (антигеннеспецифического) иммунитета и гиперпродукция острофазовых реактантов – СРБ, сывороточного амилоида А и др. Основной медиатор воспаления — ИЛ-1.
Впервые больного с периодической лихорадкой в научно-медицинской литературе представил английский клиницист У. Геберден в 1802 г [8]. Видимо, это было первое описание пациента с АВС. Однако на него не обратили внимание. Время для изучения данной проблемы тогда еще не пришло. Первое полноценное описание пациентов с семейной средиземноморской лихорадкой было сделано в 1908 г Janeway и Mosenthal, а в 1945 г. S. Siegal [9,10]. Однако и эти публикации не привлекли внимания к проблеме. Истинным началом изучения АВС можно считать 1948г., когда H. A.Reiman охарактеризовал пациентов с периодической лихорадкой, появившейся в детском возрасте и персистировавшей в течение нескольких лет и десятилетий с циклами определённой продолжительности, предложив для выделенной им патологии термин «периодическая болезнь» [11]. Значительный прогресс в изучении аутовоспалительных синдромов произошёл в последнее двадцатилетие ХХ века и был обусловлен стремительным развитием молекулярной биологии и молекулярной медицины, установившим генетическую природу указанных состояний. В 1982 г. был описан TRAPS-синдром, в 1984 г J. W.M. van der Meer гипер IgD-синдром. Список АВС был увеличен до 4 и стал включать в себя кроме семейной средиземноморской лихорадки, гипер-IgD –синдром (Hyper-Immunoglobulinemia D — syndrome – HIDS), синдром периодической лихорадки, ассоциированный с рецепторами к фактору некроза опухоли (Tumor necrosis factor Receptor-Assotiated Periodic fever Syndrome –TRAPS), и синдром Макл-Уэлса (Makle –Wells syndrome –MWS). В последующем список болезней этой группы периодически расширялся и к настоящему времени включает перечисленные выше состояния. Окончательно принятой классификации АВС нет. В настоящее время всё чаще высказывается гипотеза, что к аутовоспалительным по своей природе заболеваниям могут быть отнесены такие хорошо известные нозологии ревматологической рубрики как подагра, системная форма ювенильного артрита, болезнь Стилла у взрослых, а также синдром Висслера-Фанкони.
В данном обзоре представлены наиболее часто встречающиеся АВС, с которыми в любой момент могут столкнуться как врачи общей практики, так и узкие специалисты.
Семейная средиземноморская лихорадка (Familial Mediterranean Fever –FMF).
Исторически первой из АВС была описана семейная средиземноморская лихорадка – Familial Mediterranean Fever (FMF). FMF является самым распространённым из АВС. В мире этим синдромом страдают более 100000 пациентов. Заболевание встречается в определённых этнических группах, относящихся к народам средиземноморского бассейна. Наиболее подвержены заболеванию представители 4 этнических групп: евреев-сефардов, арабов, турок, армян [1,2]. По данным различных авторов, в основе болезни лежат мутации генов, наиболее распространёнными среди которых являются M694V и V726A и, несколько реже М680I [12,13,14,15]. Дебют заболевания у почти 75-89% пациентов с FMF относится к возрасту до 20 лет [1,2]. В табл. 3 указаны ведущие клинические проявления FMF, кроме того возможно выявление гепатоспленомегалии, затяжной фебрильной миалгии, различных нарушений со стороны сердечно-сосудистой системы, разнообразных неврологических и психоневрологических проявлений. Во время атаки заболевания у пациентов отмечается повышение острофазовах реактантов: СОЭ, сывороточных концентраций СРБ, SAA, гаптоглобина, лейкоцитоз с нейтрофилёзом, которые у 2/3 больных повышены и вне атак [1,2,3].
Основным осложнением FMF является АА-амилоидоз с преимущественным поражением почек, что и являлось в до колхициновую эру ведущей причиной гибели этих больных. Может также развиваться амилоидоз ЖКТ, печени, селезёнки, редко – сердца, яичек и щитовидной железы. Другим осложнением FMF является спаечная болезнь [2,13,14].
Диагноз FMF ставится на основании сочетания периодически повторяющихся, как правило, через определенные промежутки времени «беспричинных» лихорадочных эпизодов продолжительностью от полудня до 3-х суток, с эпизодами мучительных болей в животе с наличием симптомов раздражения брюшины, возможно, в сочетании с болями в груди и артритами у пациента характерной этнической принадлежности. Важным моментом является значительное повышение уровня острофазовых показателей во время атаки.
Для диагностики наиболее часто используются критерии Тель-Хашомера [16], которые, однако, адаптированы для популяций с высокой частотой встречаемости данного синдрома. Определённым подспорьем может служить молекулярно-генетическое типирование характерных мутаций гена MEFV.
Основой лечения FMF является колхицин. С 1972 г, когда лечение колхицином было внедрено в широкую клиническую практику, прогноз у пациентов с FMF коренным образом изменился в лучшую сторону [15]. Даже в тех случаях, когда колхицин не предотвращает рецидивы симптоматики, он значительно снижает риск развития амилоидоза. Максимальной дозой является 2 мг/сутки. Если эта дозировка не предотвращает развитие атак, её дальнейшая эскалация не имеет смысла [2,17,18]. В качестве симптоматического средства во время атак используются НПВП [2]. С развитием средств биологической терапии в лечении FMF у колхицин-резистентных пациентов с успехом стали использоваться генно-инженерные биологические препараты (ГИБП), блокирующие функции Ил-1 (анакинра) и ФНОα (инфликсимаб) [19].
Криопирин — ассоциированные периодические синдромы
(Cryopyrin Assotiated Periodic Syndromes — CAPS).
Криопирин-ассоциированные периодические синдромы (CAPS) – представляют собой группу редких врожденных аутовоспалительных заболеваний, которая включает в себя: 1. семейный холодовой аутовоспалительный синдром/семейную холодовую крапивницу (Familial Cold Autoinflammatory Syndrome/Familial Cold Urticaria –FCAS/FCU); 2. синдром Макл-Уэлса (Muckle-Wells Syndrome – MWS); 3.хронический младенческий нервно-кожно-артикулярный синдром/младенческое мультисистемное воспалительное заболевание (Chronic Infantile Onset Neurologic Cutneous Articular/Neonatal Onset Multisystem Inflammatory Disease – CINCA/NOMID).
| Из за большого объема этот материал размещен на нескольких страницах: 1 2 3 |

Источник
Kриопирин-ассоциированный периодический синдром. Что это?
Kриопирин-ассоциированный периодический синдром (КАПС) — это три редких наследственных аутовоспалительных заболевания, которые объединены мутацией в одном и том же гене и имеют схожие симптомы.
Kриопирин-ассоциированный периодический синдром включает:
1 — семейный холодовой аутовоспалительный синдром (familial cold autoinflammatory syndrome, FCAS);
2 — синдром Макла-Уэллса (Muckle Wells, MWS);
3 — мультисистемное воспалительное заболевание с неонатальным началом (neonatal onset multisystem inflammatory disease, NOMID), которое также известно как хронический инфантильный неврологический, кожный и суставный синдром (chronic infantile neurologic, cutaneous, articular, CINCA).
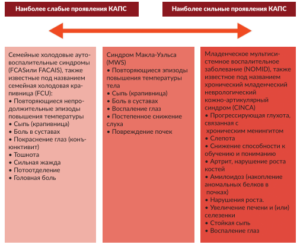
Все три заболевания значительно варьируются по степени тяжести клинических проявлений: FCAS характеризуется наиболее легким течением, синдром MWS занимает промежуточное положение, NOMID/CINCA протекает наиболее тяжело.
Kриопирин-ассоциированный периодический синдром. Причины возникновения.
 В основе развития трех фенотипов криопирин-ассоциированного периодического синдрома лежат наследственные или приобретенные de novo мутации гена NLRP3(кодирующего криопирин), которые сопровождаются неконтролируемой активацией инфламмасомы и повышением экспрессии интерлейкина (ИЛ)-1 β.
В основе развития трех фенотипов криопирин-ассоциированного периодического синдрома лежат наследственные или приобретенные de novo мутации гена NLRP3(кодирующего криопирин), которые сопровождаются неконтролируемой активацией инфламмасомы и повышением экспрессии интерлейкина (ИЛ)-1 β.
Проще говоря, нарушение функции гена ведет к усиленной выработке белка криопирина и провоцирует воспаление.
Kриопирин-ассоциированный периодический синдром. Частота встречаемости.
Kриопирин-ассоциированный периодический синдром (КАПС) — это чрезвычайно редкое заболевание. Примерно один случай на миллион человек. Но недостаточный уровень знаний большинства врачей о редких заболеваниях позволяет говорить о недостаточной диагностике, т.е. частота встречаемости КАПС должна быть намного больше. Kриопирин-ассоциированный периодический синдром диагностирован во многих странах мира. Зависимости от национальности или расы не наблюдается.
Kриопирин-ассоциированный периодический синдром. Наследственность.
Kриопирин-ассоциированный периодический синдром (КАПС) имеет аутосомно — доминантный тип наследования, т.е. заболевание передается от одного из родителей, который имеет дефектный ген. Вероятность передачи такого гена — 50 %. В некоторых случаях мутация гена у родителей не обнаруживается, вероятно генетическое нарушение возникло в момент зачатия.
Kриопирин-ассоциированный периодический синдром. Симптомы заболевания.
 Сыпь — это самый первый и самый распространенный симптом КАПС. Встречается во всех трех типах заболевания. Мигрирующая макулезно-папулезная сыпь (по типу крапивницы) имеет разную интенсивность в зависимости от типа заболевания. При NOMID/CINCA проявляется сразу после рождения, при остальных формах — в течение первого года. Кожная сыпь не зудит, возникает периодами, проходит самостоятельно.
Сыпь — это самый первый и самый распространенный симптом КАПС. Встречается во всех трех типах заболевания. Мигрирующая макулезно-папулезная сыпь (по типу крапивницы) имеет разную интенсивность в зависимости от типа заболевания. При NOMID/CINCA проявляется сразу после рождения, при остальных формах — в течение первого года. Кожная сыпь не зудит, возникает периодами, проходит самостоятельно.
Каждая отдельная форма криопирин-ассоциированного периодического синдрома имеет свои отличительные особенности и свои клинические проявления:
 FCAS, также называемый «семейная холодовая крапивница» или семейная полиморфная холодовая сыпь или холодовая гиперчувствительность, проявляется перемежающимися эпизодами сыпи, лихорадки, болями в суставах. Также повышается температура тела, наблюдаются боли в мышцах, развивается конъюнктивит.
FCAS, также называемый «семейная холодовая крапивница» или семейная полиморфная холодовая сыпь или холодовая гиперчувствительность, проявляется перемежающимися эпизодами сыпи, лихорадки, болями в суставах. Также повышается температура тела, наблюдаются боли в мышцах, развивается конъюнктивит.
Провоцируется такая клиническая картина воздействием низких температур, проще говоря, больные плохо начинают себя чувствовать после пребывания на холоде. Примерно через два часа развивается сыпь, присоединяется головная боль, лихорадка. По прошествии 12 — 24-х часов симптомы холодовой крапивницы приходят. Очевидно, что зимой проявления FCAS случаются чаще.
 MWS отмечается похожими симптомами: сыпью, температурой, периодической лихорадкой, хотя последняя бывает не всегда. Можно заметить повышенную утомляемость пациента, болезненность от воспаления суставов, воспаление глаз. MWS от FCAS отличается провоцирующими факторами: прямой зависимости от холода не наблюдается, а также наличием осложнений.
MWS отмечается похожими симптомами: сыпью, температурой, периодической лихорадкой, хотя последняя бывает не всегда. Можно заметить повышенную утомляемость пациента, болезненность от воспаления суставов, воспаление глаз. MWS от FCAS отличается провоцирующими факторами: прямой зависимости от холода не наблюдается, а также наличием осложнений.
Уже в детском возрасте у больных развивается нейросенсорная тугоухость и нередко – АА-амилоидоз с преимущественным поражением почек. Амилоидоз — отложение белка амилоида в органах и тканях, и вызывающего их поражение.
CINCA/NOMID — самая тяжелая форма КАПС. Первые проявления начинаются практически сразу после рождения. Мамы замечают у новорожденных сыпь, периодическое повышение температуры тела. Дети выглядят слабыми, часто плачут. С возрастом болезнь приводит к воспалению суставов: они болят и отекают. В особо тяжелых случаях суставы деформируются, ограничивается их подвижность. Пациент может перестать ходить.
Одно из проявлений болезни — поражение центральной нервной системы, оно связано с хроническим асептическим менингитом. Ребенок может быстро утомляться, испытывать сильные головные боли, которые сопровождаются тошнотой и рвотой. Обращают на себя внимание периодические эпилептические припадки и когнитивные нарушения.
При заболевании страдают глаза: они воспаляются, ухудшается зрение, может развиться полная слепота. Страдают уши: часто встречается нейросенрорная тугоухость. Также развивается амилоидоз.
Совокупность проявлений заболевания приводит к отставанию ребенка по всем параметрам: он сильно ниже своих сверстников, он значительно уступает им в умственном развитии, половое созревание задерживается.
Несмотря на разность проявлений симптомов у трех форм криопирин-ассоциированного периодического синдрома, четкую границу между ними в клинических случаях бывает провести довольно сложно. Иногда заболевание у конкретного человека проявляется совокупностью всех симптомов КАПС.
Криопирин-ассоциированный периодический синдром. Диагностика.
КАПС – это наследственное заболевание, первые симптомы которого практически всегда появляются в детском возрасте. Как правило, в первый год жизни появлется лихорадка и сыпь, в дальнейшем — патологические изменения нарастают. Подросший ребенок уже может жаловаться на головную боль, на боль в коленках и щиколотках.
Обычно такие дети попадают на прием к ревматологу, который подозревает аутоиммунное заболевание. Точный диагноз при КАПС ставиться гораздо позднее, примерно в 15 лет, когда отставание от сверстников видно отчетливо.
Диагностика криопирин-ассоциированного периодического синдрома (КАПС) проводится на основании клинической картины заболевания. Для подтвержения диагноза исследуют спинномозговую жидкость (люмбальная пункция), делают рентгенологическое обследование и исследование глазного дна.
При подозрении на криопирин-ассоциированный периодический синдром желательно делать генетический анализ, чтобы подтвердить наличие мутации в определенном гене.
Криопирин-ассоциированный периодический синдром. Лечение.
 Как и все редкие генетические заболевания вылечить криопирин-ассоциированный периодический синдром невозможно, но можно, введя препарат, значительно улучшить состояние больного, а вместе с тем и его жизнь.
Как и все редкие генетические заболевания вылечить криопирин-ассоциированный периодический синдром невозможно, но можно, введя препарат, значительно улучшить состояние больного, а вместе с тем и его жизнь.
Тем более, что своевременное введение препарата (при своевременной диагностике) способно минимизировать воздействие болезни на организм.
Что это за препараты? В настоящий момент их существует три. Все они ингибируют ИЛ-1β — белок, который играет ключевую роль в активации заболевания.
Анакинра (Кинерет) – это рекомбинантный негликозилированный антагонист человеческих ИЛ-1 рецепторов, который блокирует активность цитокина путем конкурентного взаимодействия с рецепторами ИЛ-1 типа I. Необходимо ежедневное подкожное введение. В Российской Федерации препарат не зарегистрирован.
Рилонацепт (Аркалист) – это химерный белок, содержащий внеклеточные домены ИЛ-1 рецептора I типа и адаптерного белка, которые присоединены к молекуле человеческого IgG. Препарат блокирует взаимодействие ИЛ-1 β с рецепторами. В Российской Федерации не зарегистрирован.
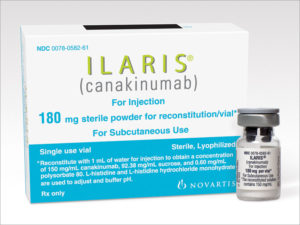
Канакинумаб (Иларис) – это человеческие моноклональные IgG1 антитела к ИЛ-1 β , которые связываются с цитокином и блокируют его взаимодействие с рецепторами. Канакинумаб обладает длительным периодом полувыведения, его нужно вводить подкожно один раз в 8 недель. Рекомендуемая стартовая доза у взрослых составляет 150 мг. Эффективность и безопасность канакинумаба проверены клиническими исследованиями. Препарат зарегистрирован и может применяться в Российской Федерации.
Лечение препаратами необходимо продолжать постоянно, пожизненно!
Криопирин-ассоциированный периодический синдром. Прогноз для жизни.
 Как показывают наблюдения за пациентами с криопирин-ассоциированным периодическим синдромом, те из них, кому начинают вводить препарат — меняются на глазах. Исчезают головные боли и лихорадка, тело больше не покрывается сыпью, а суставы не болят. Дети начинают интенсивно расти и набирать в весе. Частично компенсируются умственные способности. Ребенок может продолжать обучение в школе, общаться на равных со сверстниками и друзьями.
Как показывают наблюдения за пациентами с криопирин-ассоциированным периодическим синдромом, те из них, кому начинают вводить препарат — меняются на глазах. Исчезают головные боли и лихорадка, тело больше не покрывается сыпью, а суставы не болят. Дети начинают интенсивно расти и набирать в весе. Частично компенсируются умственные способности. Ребенок может продолжать обучение в школе, общаться на равных со сверстниками и друзьями.
При всех криопирин-ассоциированных периодических синдромах показано ведение здорового образа жизни, умеренное занятие физкультурой и спортом, сохранение душевного равновесия. Большое влияние оказывает климат, пациентам с КАПС желательно менять его на более теплый.
Источник