Аутосомно рецессивный поликистоз почек синдром


Поликистозная болезнь почек с аутосомно-рецессивным типом наследования (ПБП-АР) принадлежит к группе врожденных гепаторенальных фибрознокистозных заболеваний. Первично всегда поражаются почки и печень, но с развитием заболевания в патологический процесс могут включаться и другие органы. Заболевание характеризуется двусторонним увеличением почек из-за наличия небольших множественных кист и поражением печени.
Эпидемиология
Младенческая форма. Встречается редко (1:10000–1:40000), частота может расти в изолированных популяциях. На данный момент не выявлено существующих гендерных и расовых предрасположенностей.
Этиология
Несмотря на клиническую вариабельность и множественное поражение органов, только единичная мутация гена PKHD1 в 6 хромосоме (6p12) отвечает за заболевание. Ген PKHD1 отвечает за экспрессию белка фиброцистина преимущественно в печени, почках и поджелудочной железе. Белок фиброцистин входит в состав первичных ресничек, располагающихся на апикальной мембране клеток эпителия почек и желчных путей.
Классификация
В зависимости от времени начала первых проявлений и степени поражения печени выделяют следующие типы заболевания:
1. Перинатальный тип — наиболее распространенный:
- сопровождается маловодием и гипоплазией легкого;
- 75 % умирают в течение 24 часов после родов;
- минимальная степень фиброза печени;
2. Неонатальный тип — минимальная степень перипортального фиброза печени;
3. Младенческий тип — умеренный перипортальный фиброз;
4. Ювенильный тип — выраженный перипортальный фиброз в сочетании с портальной гипертензией, спленомегалией и портосистемным варикозом.
Патологическая анатомия
Макроскопическое строение
Плод имеет характерный внешний вид (лицо Поттера): узкие щели век, характерная борозда под линией века, микрогнатия, приплюснутый нос, мягкие большие уши аномальной формы. Почки увеличены в размерах, при этом сохраняют свою бобовидную форму, но не могут выполнять свою функцию. Плохо функционирующие почки не производят достаточное количество фетальной мочи, что приводит к маловодию и гипоплазии легких. Гидростатическое давление амниотической жидкости обеспечивает нормальное развитие дыхательной системы.
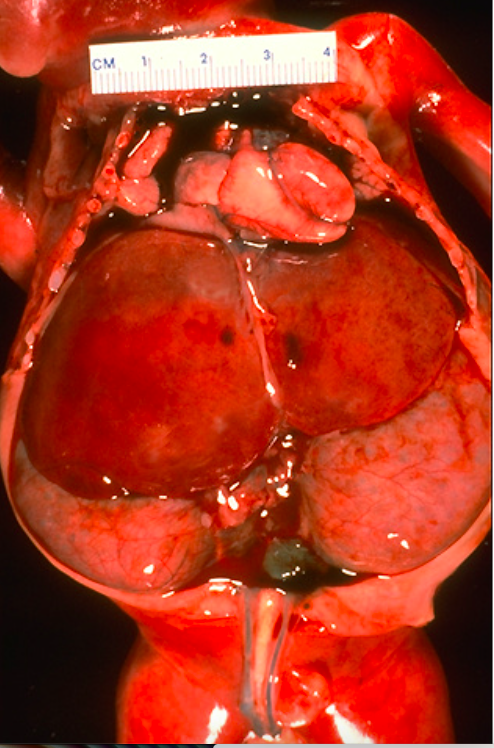
Рисунок 1 | Плод, 23 недели. Смерть плода после преждевременных родов наступила из-за гипоплазии легких, причиной которой является маловодие вследствие ПБП.
Увеличение обеих почек со смещением органов брюшной полости в сочетании с гипоплазией легкого.
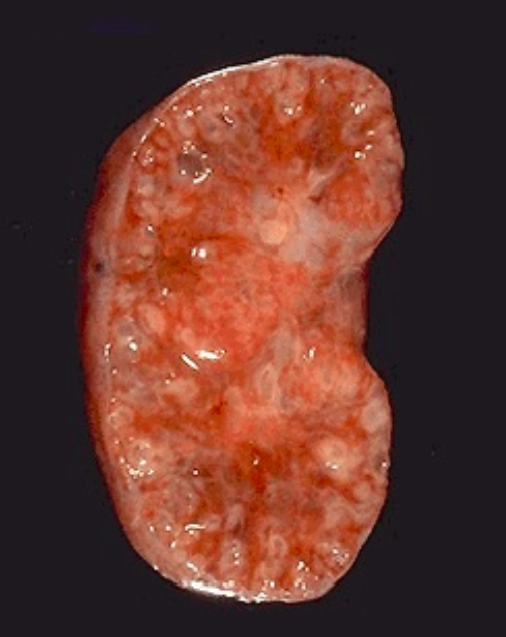
Рисунок 2 | Продольный срез почки с ПКБ.
Микроскопия
Почки
Радиально ориентированные расширенные собирательные канальцы формируют почечные кисты диаметром 1–2 мм, между которыми видны нормальные клубочки и канальцы. Размеры кист могут варьировать в зависимости от возраста. На ранних стадиях болезнь дебютирует с микрокист, которые затем растут и превращаются в макрокисты. Кистозное поражение почек сопровождается незначительным интерстициальным фиброзом паренхимы почек.
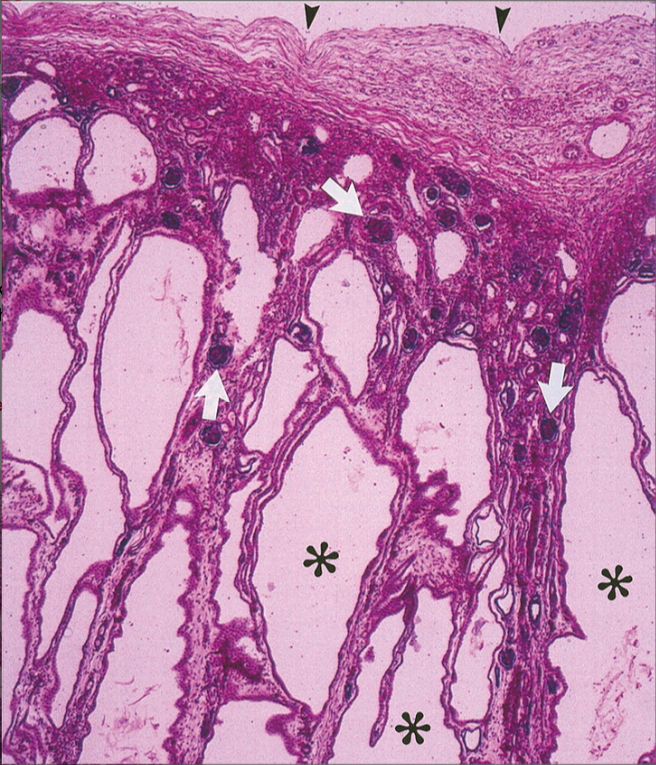
Рисунок 3 | Микропрепарат почки пациента с ПБК-АР. Двадцатикратное увеличение, окраска гематоксилин-эозин.
✱ — радиальные почечные кисты;
▼— почечная капсула.
Стрелками обозначены нормальные клубочки между расширенными собирательными канальцами.
Печень
Гистологические изменения печени включают следующие пороки развития дуктальной пластинки: гиперплазию желчевыводящих путей, билиарную эктазию и перипортальный фиброз. Нарушения морфогенеза развития желчных путей приводит к их дилатации. С последующим прогрессированием заболевания расширенные протоки превращаются в макрокисты, связанные нормальными протоками, что позволяет достаточно хорошо их верифицировать с помощью МРХПГ.
Рисунок 4 | Патологические изменения печени.
a) Строение нормально разветвленной портовенозной и решетчатой систем желчных путей (слева) нарушается вследствие дефекта развития дуктальной пластинки и ошибок в терминальной дифференцировке холангиоцитов (справа);
b) Корональное Т2-взвешенное изображение брюшной полости:
— стрелкой показано кистозное веретенообразное расширение желчных протоков;
— стрелкой по типу наконечника стрелы — нефромегалия с маленькими цистами;
✱ — спленомегалия.
c) Препарат фрагмента печени, окраска гематоксилин-эозин, увеличение в 40 раз:
✱ — обширный фиброз портальной области;
стрелка — расширенные извилистые желчные протоки;
стрелка по типу наконечника стрелы — гипоплазия притоков портальной вены.
Формирование кист
При ПБП-АР кисты формируются преимущественно из дистальных канальцев и собирательных трубочек, детальные молекулярные механизмы этого до сих остаются покрыты мраком. В результате неправильного восприятия сигналов из-за нарушенной работы первичных ресничек в клетках почечного эпителия происходит нарушение внутренних сигнальных механизмов пролиферации и дифференцировки, что приводит к образованию пузырьков, наполненных жидкостью.
Клиническая характеристика
Первые симптомы у большинства пациентов проявляются в неонатальном периоде.
Почки
При осмотре: увеличение правых и левых боковых областей живота, где пальпируются объемные образования. Объем выделяемой мочи обычно уменьшен, хотя при нарушении концентрационной функции почек могут иметь место явления полиурии и полидипсии. Тяжелые формы с развитием острой почечной недостаточности (ОПН) будут проявляться олигурией, гипонатриемией, увеличением креатинина и азота мочевины в крови. Но с возрастом благодаря развитию нормальной почечной ткани происходит улучшение почечной функции и уменьшение проявлений ОПН. Несмотря на это, примерно у 50 % пораженных в первую декаду жизни болезнь прогрессирует в терминальную почечную недостаточность. Симптомы тяжелой артериальной гипертензии, которая дебютирует с первых дней, также становятся менее тяжелыми с течением времени.
Печень
С развитием методов заместительной терапии функций почек и трансплантологии улучшается долгосрочная выживаемость пациентов. Благодаря этому гепатобилиарные клинические проявления становятся основной проблемой старших возрастных групп. Врожденный фиброз печени приводит к прогрессирующей портальной гипертензии, что проявляется варикозно расширенными венами пищевода, желудка, геморроидальных вен, гепатоспленомегалией, энтеропатией с потерей белка и желудочно-кишечными кровотечениями.
Синдром Кароли— необструктивная дилатация внутрипеченочных желчных протоков и общего желчного протока, встречающаяся более чем в 60 % случаев ПКБ-АР. Недостаточность дренажной функции желчной системы предрасполагает к развитию бактериального восходящего холангита и сепсиса. Мальабсорбция из-за холестаза приводит к дефициту жирорастворимых витаминов (A, D, E и K). У взрослых пациентов гиперплазия желчевыводящих путей предрасполагает к возникновению добро- и злокачественных опухолей.
Диагностика
Диагноз ПБП-АР должен быть заподозрен во всех случаях диффузного эхогенного увеличения обеих почек при УЗИ. Для постановки точного диагноза обычно бывает достаточно клинических признаков и наличия радиографических изменений. Но для точной верификации заболевания разработаны специальные диагностические критерии: сочетание патогномоничных признаков изменения почки — одного или нескольких пунктов из следующих:
- эктазия желчных протоков (см. ультрасонографию);
- клинические и лабораторные признаки врожденного фиброза печени (ВФП);
- гепатобилиарная патология, характерная для аномалий развития дуктальной пластины и сопутствующая ВФП;
- наличие морфологических (биопсия или аутопсия) и генетических признаков.
Ультразвуковое исследование
УЗИ высокого разрешения значительно улучшило диагностику легких форм заболевания, когда клинические проявления невыраженные, обеспечивая неинвазивную детальную визуализацию изменений в почках без использования радиации и контрастных веществ.
Ультрасонография является методом выбора при исследовании плода и детских форм ПКБ-АР. УЗИ диагностика обладает большим перечнем преимуществ: низкая стоимость, безболезненность, широкая распространенность и отсутствие необходимости в излучении и седации. Но несмотря на все перечисленные достоинства УЗИ, с целью верификации диагностика дополняется МРТ.
Патогномоничные изменения почек, диагностируемые при помощи УЗИ:
- увеличение размера почек (с учетом возрастных особенностей);
- повышенная эхогенность;
- снижение кортико-медуллярной дифференциации.
Внутриутробно:
- обнаруживаются увеличенные бобовидные почки со сглаженной кортико-медуллярной дифференциацией, маловодие и пустой мочевой пузырь в некоторых случаях;
- сильно выраженное маловодие приводит к легочной гипоплазии и высокой смертности вследствие легочной недостаточности. Помимо этого, олигоамнион может сочетаться с множественными аномалиями лица, конечностей и других органов по типу синдрома Поттера.
Младенчество (до года):
- наличие пальпируемых объемных образований в обеих боковых областях живота с идиопатической хронической легочной патологией, маловодие в анамнезе матери или спонтанный пневмоторакс у новорожденного и артериальная гипертония позволяют предположить ПБП-АР;
- сочетание аномалий билиарной системы вместе с признаками портальной гипертензии, такими как гепатоспленомегалия, добавляет уверенности для постановки диагноза ПБП-АР.
Детство и юношеский возраст:
- с возрастом размер и степень фиброза почек будут прогрессировать вместе с аномалиями гепатобилиарного тракта, что приводит к прогрессирующей портальной гипертензии;
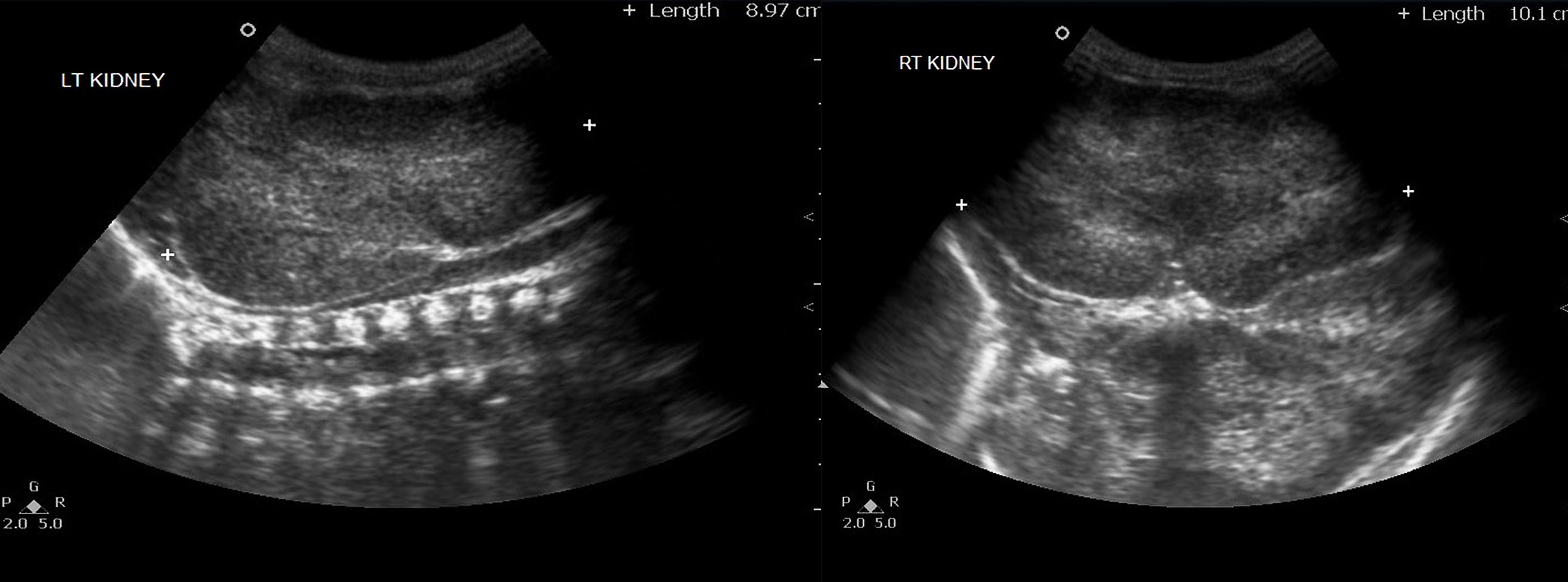
Рисунок 5 | Обе почки увеличены в продольном размере (правая 10 см и левая 9 см), с повышенной эхогенностью (за счет акустического усиления крошечных кист) и измененной кортикомедуллярной дифференцировкой.
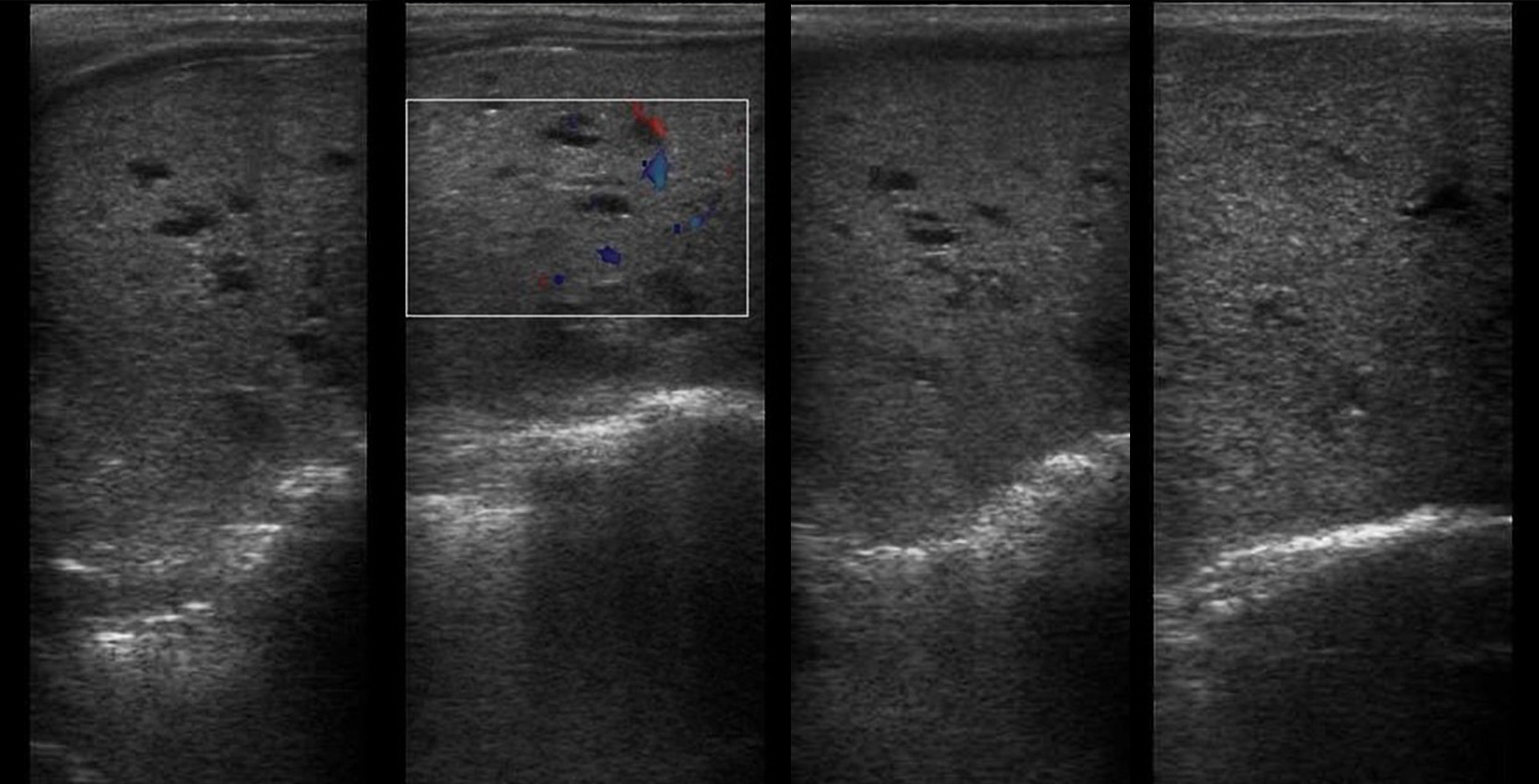
Рисунок 6 | Печень с врожденным фиброзом и кистозными расширениями в правой доле.
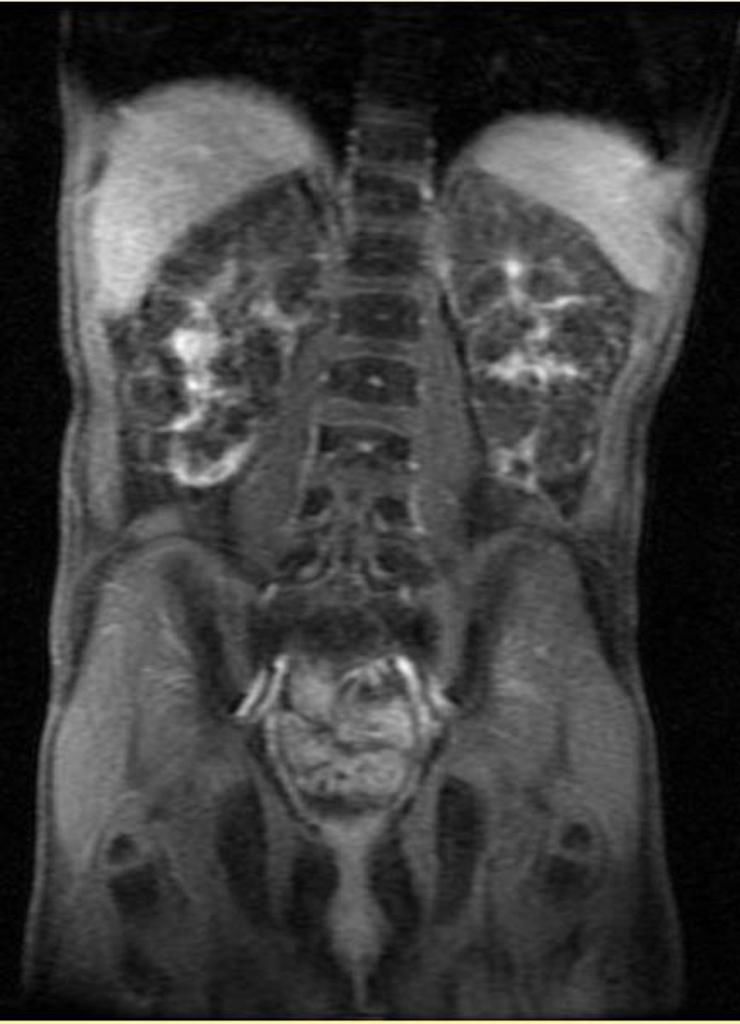
Рисунок 7 | МРТ, корональная Т2-проекция, пациент с ПБП-АР.
МРТ
Магнитно-резонансный томограф не имеет никаких преимуществ перед ультразвуковыми сканерами высокого класса и генетическим тестированием в диагностике ПКП-АД.
Магнитно-резонансная холангиопанкреатография (МРХПГ)
Изображения, получаемые с помощью МРХПГ, обеспечивают хорошее качество визуализации гепатобилиарной системы у пациентов с ПБП-АР. МРХПГ является чувствительным методом диагностики билиарной эктазии, который в сочетании с визуализацией почек почти полностью заменил такие инвазивные методы диагностики, как биопсия почек и печени.
NB! Биопсия почек и печени не должны использоваться для диагностики ПБП.
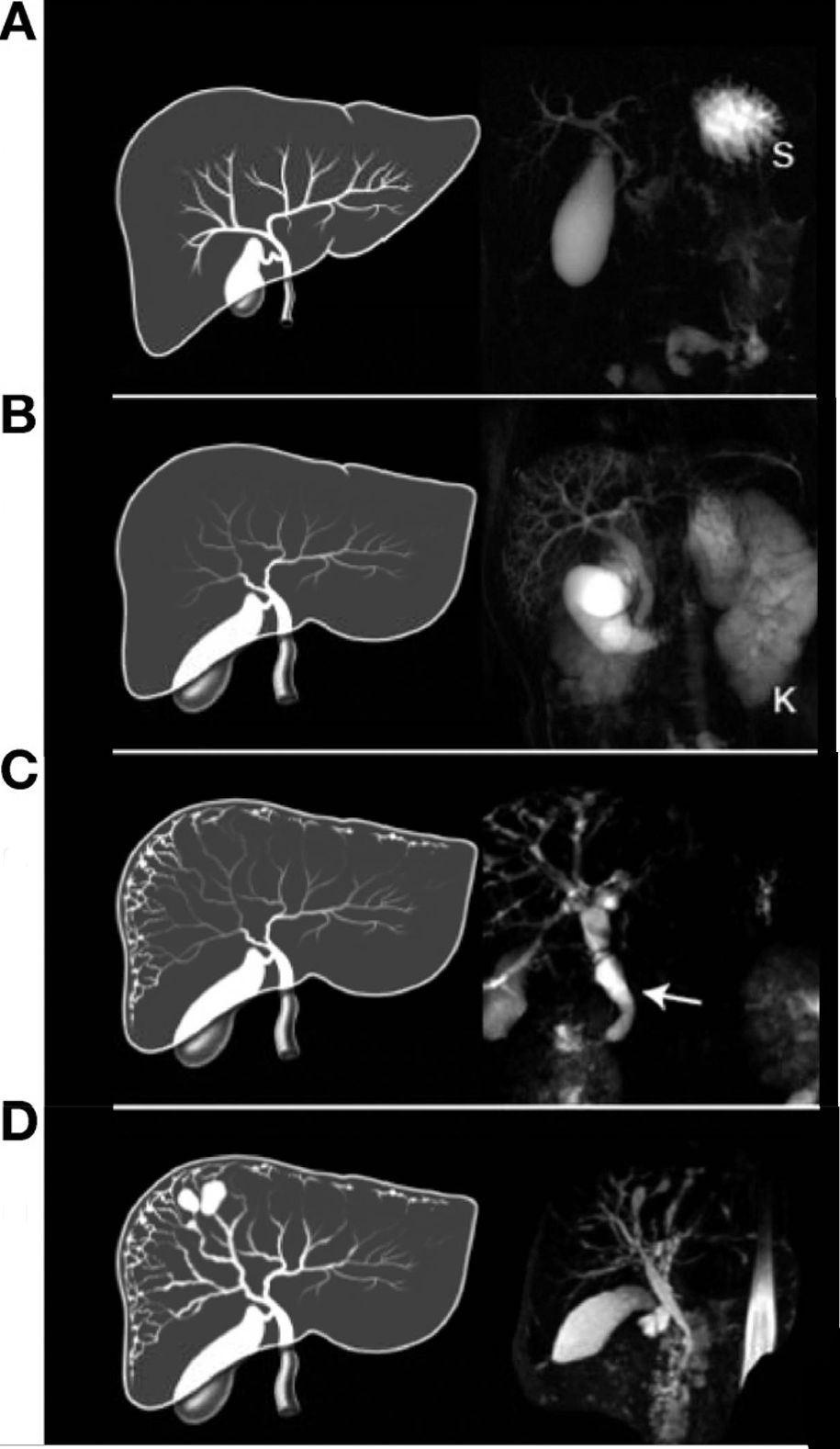
Рисунок 8 | МРХПГ и последующий рендеринг билиарной системы с аномалиями, свойственными для пациентов с ПБП-АР:
A. Нормальная печень с увеличенным желчным пузырем, (S) желудок.
B. Увеличенный общий желчный проток с желчным пузырем. Расширенные внутрипеченочные протоки.
C. Веретенообразные кисты периферических и центральных желчных протоков небольшого размера с расширенным общим желчным протоком (стрелка) и желчным пузырем.
D. Веретенообразные макроцисты с расширенными печеночными протоками.
У всех пациентов была печень аномальной формы с непропорционально увеличенной левой долей.
Дифференциальная диагностика
ПБП-АР следует дифференцировать со следующими состояниями:
- аутосомно-доминантная поликистозная болезнь почек;
- почечная дисплазия, связанная с трисомией 13 хромосомы;
- синдром Ме́ккеля — Гру́бера;
- синдром Лоренса — Муна — Барде — Бидля;
- синдром Беквита — Видемана;
- гломерулярная кистозная (гломерулокистозная) болезнь почек.
Подтверждение диагноза
Если имеются серьезные подозрения на ПБП-АР, необходим генетический анализ и консультирование. Молекулярно-генетическое тестирование показано пробанду с наличием у него кистозного увеличения почек и ВФП для выявления обеих патологических аллелей PKHD1. Должны быть проанализированы как минимум 3 поколения, т. к. заболевание передается рецессивно. Молекулярно-генетическое подходы к тестированию включают в себя панели для диагностики единичных точечных мутаций, мультигенные панели или полное геномное исследование.
Лечение
Аппарат искусственного кровообращения необходим всем пациентам с гипоплазией легкого, у которых оксигенация 100 % кислородом неэффективна из-за гипервентиляции вследствие давления на диафрагму. Новорожденным с врожденной легочной недостаточностью может потребоваться ИВЛ с первых минут жизни до определения причины. Когда увеличенные почки сдавливают ЖКТ, ряд авторов рекомендуют двух- или одностороннюю нефрэктомию, но при односторонней нефрэктомии может наблюдаться компенсаторное еще большее увеличение оставшейся почки. После двусторонней нефрэктомии пациенты нуждаются в гемодиализе.
Хронический перитонеальный диализ, с короткими периодами на гемодиализ для восстановления брюшины — метод выбора. Продолжительность этих процедур, также как и превентивная трансплантация почки, будут зависеть от ряда таких факторов как возраст, вес, клинический статус пациента и наличие здорового подходящего донора. Лечение гипонатриемии должно проводиться в зависимости от степени обезвоживания — раннее распознавание и лечение дегидратации имеют решающее значение. Для введения дополнительного питания и жидкостей может потребоваться установка назогастрального зонда. Артериальная гипертония (АГ) обычно хорошо поддается лечению ингибиторами АПФ и блокаторами рецепторов ангиотензина, которые являются препаратами выбора. Порой АГ может потребовать сочетания нескольких препаратов. Для лечения анемии у детей с хронической почечной недостаточностью необходимо добавление препаратов железа или стимуляторов выработки эритропоэтина. Лечение билиарной дисфункции должно быть сфокусировано на мальабсорбции питательных веществ и жирорастворимых витаминов и снижении риска восходящего холангита. Варикозно расширенные вены пищевода можно клипировать или использовать склеротерапию. Прогрессирующая портальная гипертензия требует портосистемного шунтирования. Благодаря возможностям современной медицины, в тяжелых случаях портальной гипертензии, почечной и печеночной недостаточности стала возможна двойная почечно-печеночная трансплантация.
Источники:
- Characteristics of Congenital Hepatic Fibrosis in a Large Cohort of Patients With Autosomal Recessive Polycystic Kidney Disease Gunay–Aygun, Meral et al. Gastroenterology , Volume 144 , Issue 1 , 112 — 121.e2
- Sweeney WE, Avner ED. Polycystic Kidney Disease, Autosomal Recessive. 2001 Jul 19 [Updated 2019 Feb 14]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1326/
- https://ghr.nlm.nih.gov
- https://emedicine.medscape.com
- https://www.msdmanuals.com
- https://www.niddk.nih.gov
- https://radiopaedia.org
- https://www.humpath.com
- https://webpath.med.utah.edu
Источник
Аутосомно-рецессивный поликистоз почек у детей. Диагностика и лечение
Многие врожденные аномалии мочевых путей могут сопровождаться макро- или микрогематурией. Внезапная макрогематурия после небольшой травмы поясничной области характерна для обструкции лоханочно-мочеточникового сегмента или поликистоза почек.
Аутосомно-рецессивный поликистоз почек — это заболевание, называемое также поликистозной болезнью младенцев, встречается с частотой 1:10000-1:40 000. Ген, ответственный за аутосом-но-рецессивный поликистоз почек, пока не найден, но исследования по генетическому сцеплению позволяют локализовать его на коротком плече хромосомы 6.
Патоморфология аутосомно-рецессивного поликистоза почек. Обе почки резко увеличены, содержат много кист в корковом и мозговом веществе. Под микроскопом видны многочисленные кисты, распространяющиеся от мозгового вещества в корковое и расположенные преимущественно в собирательных трубочках и протоках. У плодов наблюдаются преходящие кисты и в проксимальных канальцах. Прогрессирующий фиброз интерстициальной ткани и атрофия канальцев в конце концов приводят к почечной недостаточности.
Поражение печени характеризуется пролиферацией и эктазией желчных протоков, а также фиброзом, неотличимым от врожденного фиброза печени или болезни Кароли (расширение внутрипеченочных протоков с холелитиазом, рецидивирующим холангитом и желтухой).
Клинические проявления аутосомно-рецессивного поликистоза почек. В типичных случаях у новорожденных или грудных детей обнаруживаются объемные образования в боковых отделах живота. Аутосомно-рецессивный поликистоз почек может сопровождаться маловодием, гипоплазией легких, респираторным дистресс-синдромом и спонтанным пневмотораксом в неонатальном периоде. Возможны аномалии лицевого скелета (лицо Поттера) и другие последствия уменьшения объема околоплодных вод, включая низко посаженные уши, микрогнатию, уплощенный нос, контрактуру конечностей и дефицит роста.
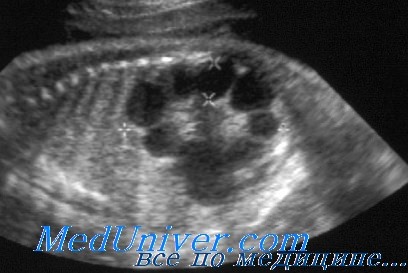
УЗИ при поликистозе почек
В первые несколько недель жизни обычно развивается высокая артериальная гипертония. Объем мочи, как правило, не снижен, хотя иногда отмечается олигурия и острая почечная недостаточность. В неонатальном периоде обычно имеет место преходящая гипонатриемия (часто на фоне острой почечной недостаточности), которую можно снять диуретиками. У 20-30 % больных функция почек остается нормальной. В редких случаях аутосомно-рецесивный поликистоз почек проявляется почечной недостаточностью, артериальной гипертонией или гепатоспленомегалией (вследствие фиброза печени) в более позднем возрасте.
Лучевые или лабораторные методы исследования определяют поражение печени примерно у 45 % новорожденных с этим заболеванием. Однако у больных повышен риск развития 1) восходящего холангита, варикозного расширения вен и гиперспленизма из-за портальной гипертензии и 2) прогрессирующей дисфункции печени, которая может привести к явной печеночной недостаточности и циррозу.
У части старших детей с аутосомно-рецессивным поликистозом почек заболевание проявляется именно гепатоспленомегалией, а поражением почек обнаруживается случайно при рентгенографии брюшной полости.
Диагностика аутосомно-рецессивного поликистоза почек. Пальпируемое двустороннее объемное образования в боковых отделах живота у грудного ребенка с гипоплазией легких, маловодием и артериальной гипертонией в отсутствие поликистоза почек у родителей позволяет диагностировать болезнь. При УЗИ почки обычно резко увеличены и равномерно повышена их эхогенность со стертостью границы между корковым и мозговым веществом. Диагноз подтверждают также клинические и лабораторные признаки фиброза печени, патологические изменения желчных протоков в биоптате печени, наличие поликистоза почек у сиблингов или близкое родство родителей. Аутосомно-рецессивный поликистоз почек следует отличать от увеличения почек при поликистозной дисплазии, гидронефроза, опухоли Вильмса и двустороннем тромбозе почечных вен. В семьях хотя бы с одним больным ребенком возможна пренатальная диагностика с помощью анализа генетического сцепления и использования информативных маркеров.
Лечение аутосомно-рецессивного поликистоза почек симптоматическое. Гипоплазия легких и гиповентиляция в неонатальном периоде часто требуют ИВЛ. Необходима нормализация АД и водно-электролитного баланса, устранение клинических проявлений почечной недостаточности. При тяжелой дыхательной недостаточности и в случаях, когда увеличенные почки препятствуют усвоению пищи, может потребоваться нефрэктомия с последующим диализом.
Прогноз аутосомно-рецессивного поликистоза почек. Около 30 % больных погибает в неонатальном периоде от осложнений гипоплазии легких. Однако, если дети не погибают в первой год жизни, то современные методы лечения дыхательной и почечной недостаточности у новорожденных увеличивают 10-летнюю выживаемость до 80 % и более. Терминальная стадия ХПН (обычно в первые 10 лет жизни) наблюдается более чем в 50 % случаев. Стандартные методы лечения детей — диализ и трансплантация почки. Последующие заболеваемость и смертность связаны с осложнениями ХПН и поражением печени.
— Также рекомендуем «Аутосомно-доминантный поликистоз почек у детей. Диагностика и лечение»
Оглавление темы «Болезни почек у детей»:
- Быстропрогрессирующий (некротический экстракапиллярный) гломерулонефрит. Диагностика и лечение
- Болезнь Гудпасчера. Гемолитико-уремический синдром
- Диагностика и лечение гемолитико-уремического синдрома. Прогноз
- Тромбоз почечных вен у детей. Диагностика и лечение
- Идиопатическая гиперкальциурия у детей. Нефропатия при серповидно-клеточной анемии
- Аутосомно-рецессивный поликистоз почек у детей. Диагностика и лечение
- Аутосомно-доминантный поликистоз почек у детей. Диагностика и лечение
- Гематурия при цистите и уретрите у детей. Диагностика и лечение
- Протеинурия у детей. Ортостатическая (постуральная) протеинурия
- Клубочковая и канальцевая протеинурия у детей. Диагностика
Источник