Асимметрия крыла носа характерна для синдрома аперта

Что такое синдром Аперта?
Синдром Аперта (синдром Апера или акроцефалосиндактилия 1 типа) — редкое генетическое заболевание, возникающее при рождении. У больных с синдромом Аперта появляются характерные пороки развития черепа, лица, рук и ног.
Синдром Апера характеризуется краниосиностозом, состоянием, при котором фиброзные суставы (швы) между костями черепа преждевременно закрываются. Это может вызвать поражение лицевых костей и привести к тому, что верхняя часть головы будет выглядеть заостренной. При болезни может наблюдаться сращивание пальцев рук или ног. Пострадавшие дети также могут иметь интеллектуальные нарушения. Серьезность симптомов варьируется.
Синдром Аперта почти всегда является результатом новых генетических изменений (мутаций), которые происходят случайно. Редко болезнь наследуется по аутосомно-доминантному типу. Больные с синдромом могут проходить терапию, направленную на конкретные симптомы, включая реконструктивную хирургию черепа, лица, рук, ног.
Признаки и симптомы
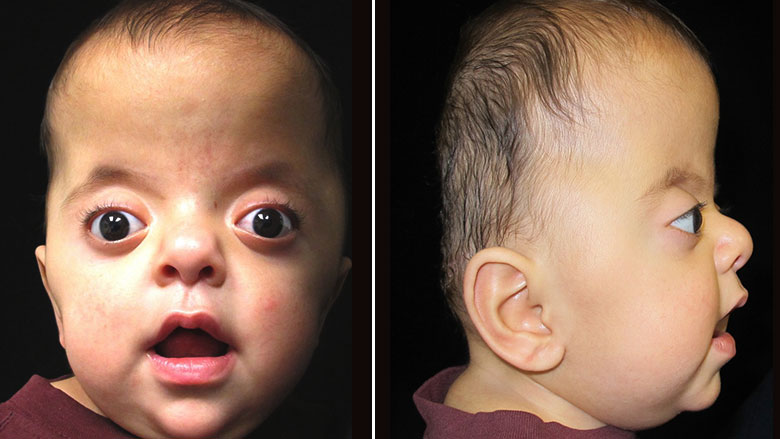
Синдром Аперта характеризуется краниосиностозом, преждевременным закрытием фиброзных суставов (швов) между определенными костями черепа. У детей без краниосиностоза швы позволяют голове расти и расширяться. Затем, эти кости сливаются воедино, образуя череп.
У людей с краниосиностозом мозг все еще растет после преждевременного закрытия этих швов.
Возникающее давление при росте головного мозга может привести к тому, что различные кости черепа и лица деформируются во время развития.
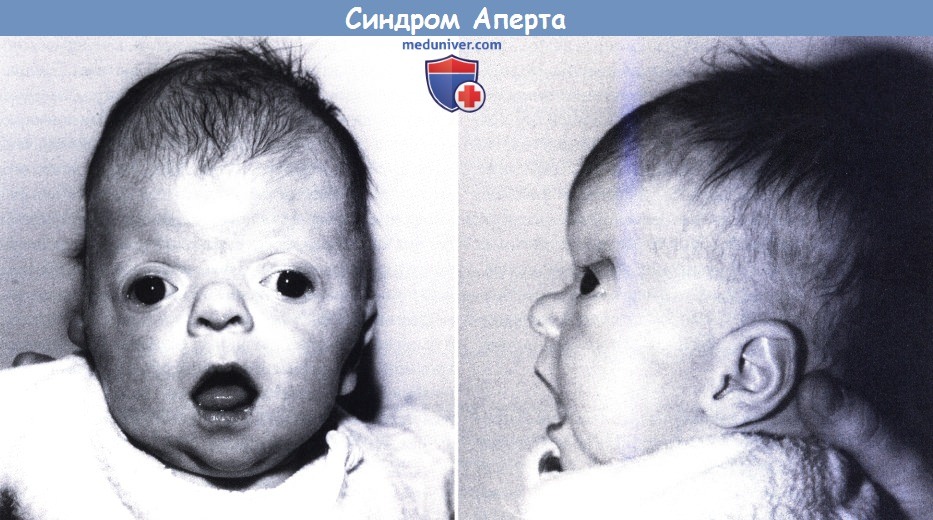
В зависимости от того, какие швы преждевременно закрываются, варьируется степень их тяжести. У большинства больных людей происходит преждевременное закрытие швов между костями, образующими лоб и верхнюю часть черепа. Это приводит к тому, что голова с рождения становится заостренной на верхней части (акроцефалия). Вдобавок, задняя часть черепа может выглядеть сплющенной, с высоким и широким лбом (см. фото выше). На черепе может быть большой «родничок» с поздним закрытием.
У некоторых больных также может быть гидроцефалия, при которой в полостях головного мозга накапливается спинномозговая жидкость. Это может вызвать внутричерепное давление.
Лицевые кости тоже могут быть поражены краниосиностозом. Это может привести к характерным дефектам лица. У пациентов с синдромом Аперта часто наблюдаются широко расставленные глаза (гипертелоризм), выпученные глаза (экзофтальм) или наклоненные вниз глазные щели. У них также могут быть слаборазвитые срединно-лицевые области (гипоплазия верхней челюсти) и расщелина нёба. Правая и левая стороны лица могут быть не симметричными.
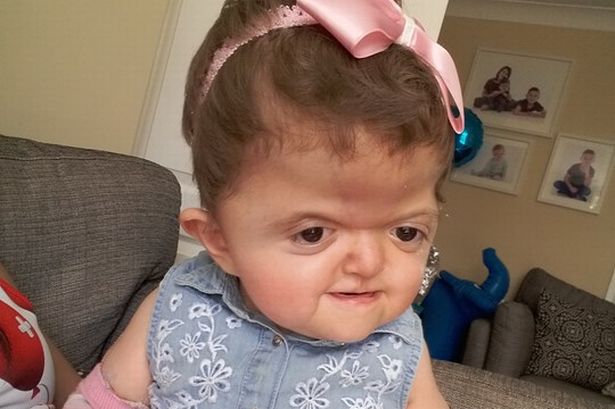
Люди с болезнью могут иметь сплющенный нос с низкой переносицей. У людей бывает задержка роста зубов, скученность зубов или открытый прикус.
Если отверстия между носом и горлом сужены или заблокированы, или трахеальный хрящ поврежден, это может помешать дыханию и глотанию. Люди с этими проблемами могут иметь инфекции верхних дыхательных путей, апноэ во сне, недоедание.
Синдром Аперта имеет несколько характерных пороков развития кисти и стопы. У пострадавших людей могут быть короткие, широкие и большие пальцы ног, отклоняющиеся наружу. Они также могут иметь частичное или полное слияние (синдактилию) определенных пальцев рук и ног. Многие больные люди имеют полное слияние костей второго и четвертого пальцев и один единственный сплошной ноготь («варежкообразная» синдактилия). Однако возможны и другие слияния.
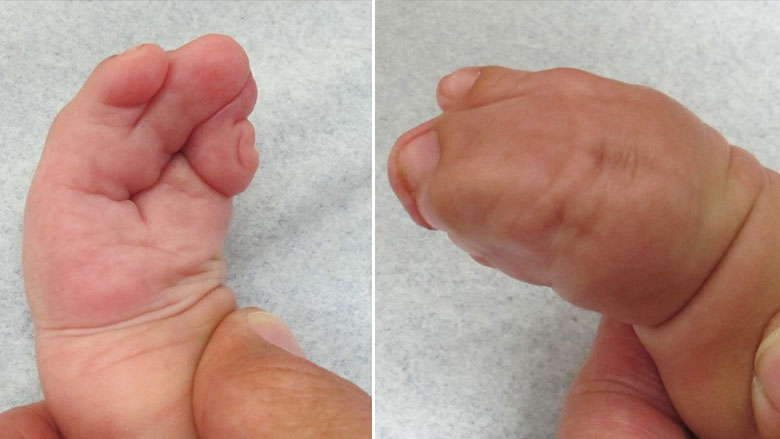
Суставы пальцев становятся жесткими к четырем годам. В ногах синдактилия также обычно затрагивает вторые, третьи и четвертые пальцы. Ногти на ногах могут быть частично непрерывными или отдельными. Как правило, синдром поражает верхние конечности сильнее, чем нижние.
Синдром Аперта может влиять и на другие системы органов (см. таблицу).
| Скелетная система |
|
| Нервная система |
|
| Уши |
|
| Сердечно-сосудистая система |
|
| Брюшная полость |
|
| Почки и мочеполовая система |
|
Причины
Синдром Аперта возникает в результате изменения (мутации) гена рецептора фактора роста фибробластов 2 (англ. Fibroblast growth factor receptor 2, сокр. FGFR2). Этот ген играет важную роль в развитии скелета. Гены предоставляют инструкции для создания белков, играющих разные роли в организме. Когда происходит мутация гена, белковый продукт может не работать должным образом. При синдроме Апера мутации в FGFR2 приводят к тому, что эти рецепторы неправильно связываются с факторами роста фибробластов. Это влияет на формирование нормальных швов в мозге и может препятствовать развитию многих других структур в организме. Это неправильное образование является причиной пороков развития, наблюдаемых при синдроме Аперта.
Почти у всех зарегистрированных пациентов расстройство было вызвано одной из двух специфических мутаций гена FGFR2. (Эти мутации обозначены как «Ser252Trp» и «Pro253Arg».) Эти мутации могут вызывать слегка отличающиеся проявления, включая степень выраженности синдактилии (сращивание пальцев). Различные мутации в гене FGFR2 могут вызывать несколько других связанных расстройств, включая синдром Пфайффера, синдром Крузона и синдром Джексона-Вейсса.
У 95% пациентов синдром Апера возникает в результате новой мутации в гене FGFR2. Эти новые мутации появляются случайно по неизвестным причинам (спорадически). Сообщалось, что отдельные случаи заболевания связаны с увеличением возраста отца.
Редко синдром наследуется по аутосомно-доминантному типу. Доминантные генетические нарушения возникают, когда необходима только одна копия мутации, чтобы вызвать конкретное заболевание. Риск передачи мутации от больного родителя к потомству составляет 50% для каждой беременности. Риск одинаков для мужчин и женщин.
Затронутые группы населения
Синдром Аперта, по оценкам, встречается примерно у одного из 65 000 новорожденных. Мужчины и женщины болеют в относительно равных количествах. С тех пор, как расстройство было первоначально описано в 1894 и 1906 годах было зарегистрировано более 300 новых случаев. Сообщалось, что у людей из Азии самый высокий уровень заболеваемости данным синдромом.
Диагностика
Диагноз чаще всего ставится при рождении или в младенчестве. Пациенты диагностируются посредством клинической оценки и различных специализированных тестов. Определяются физические признаки, такие как аномалии лица или синдактилия.
Скелетные аномалии и врожденные пороки сердца обнаруживаются с помощью визуализации, например компьютерной томографии (КТ) или магнитно-резонансной томографии (МРТ). Нарушение слуха может быть обнаружено во время исследования слуха у новорожденного.
Люди могут также пройти тест на мутации в гене FGFR2, что может обеспечить генетическим диагнозом синдрома Апера.
В некоторых случаях признаки синдрома Аперта могут быть обнаружены до рождения. Это можно сделать с помощью пренатального 2D или 3D ультразвукового исследования или магнитно-резонансной томографии (МРТ). Ультразвуковое исследование (УЗИ) — неинвазивная процедура, позволяющая получить изображение плода. Процедура может обнаружить различия в форме черепа, аномалии лица и синдактилию. МРТ плода может обеспечить большую детализацию мозга плода, чем УЗИ.
Схожие по симптомам расстройства
Симптомы следующих расстройств могут быть похожи на симптомы синдрома Аперта. Сравнения могут быть полезны для дифференциальной диагностики.
- Синдром Карпентера — редкое генетическое заболевание, связанное с краниосиностозом, полидактилией или синдактилией. Макушка головы может казаться необычно заостренной (акроцефалия) или голова может казаться короткой и широкой (брахицефалия). Вдобавок, черепные швы часто сливаются неравномерно, в результате чего голова и лицо кажутся асимметричными от одной стороны к другой. В некоторых случаях присутствуют дополнительные физические отклонения, такие как низкий рост, врожденные пороки сердца, легкое или умеренное ожирение, пупочная грыжа или крипторхизм. Многие люди с расстройством страдают от легкой до умеренной умственной отсталости.
- Синдром Крузона — редкое наследственное заболевание, связанное с краниосиностозом. У людей с синдромом Крузона также имеются пороки развития средней части лица, выпуклые глаза и закупорка дыхательных путей, приводящие к затруднению дыхания и глотанию. У некоторых больных голова очень большая (гидроцефалия). Синдром Крузона обычно не связан с умственной отсталостью или проблемами затрагивающими руки, ноги, кисти или ступни. Синдром Крузона вызывается изменениями в одном из генов FGFR, обычно FGFR2, и наследуется по аутосомно-доминантному типу.
- Синдром Джексона-Вейсса (СДВ) — редкое наследственное заболевание, характеризующееся краниосиностозом и аномалиями ног. Диапазон и серьезность симптомов и признаков сильно различаются, даже среди затронутых членов одной семьи. Первичные признаки могут включать необычно плоские, недоразвитые срединно-лицевые области (гипоплазия средней части лица), аномально широкие большие пальцы ног и/или порок развития или слияние определенных костей в ступнях. СДВ может возникать спорадически или наследоваться по аутосомно-доминантному типу.
- Синдром Пфайффера — редкое генетическое заболевание, характеризующееся краниосиностозом и аномально широкими и отклоненными медиально большими пальцами рук и ног. У большинства пострадавших также наблюдаются выпученные глаза и кондуктивная потеря слуха. Существует три формы синдрома Пфайффера. Типы II и III являются более серьезными. Синдром Пфейффера является аутосомно-доминантным расстройством, связанным с мутациями в генах FGFR2 и FGFR1.
Лечение
Лечение синдрома Аперта варьируется в зависимости от того, какие симптомы наблюдаются у больного. Лечение может потребовать ухода со стороны группы медицинских специалистов, включая педиатров и хирургов, нейрохирургов, врачей, специализирующихся на заболеваниях скелета, суставов и мышц (ортопедов), врачей, специализирующихся на заболеваниях ушей, носа и горла (отоларингологов), врачей, специализирующихся на нарушениях сердечной деятельности (кардиологов).
Специальные методы лечения синдрома Аперта являются симптоматическими и поддерживающими. Краниосиностоз и гидроцефалия могут привести к ненормально повышенному давлению внутри черепа и мозга. В таких случаях для коррекции краниосиностоза может быть рекомендовано раннее хирургическое вмешательство (в течение 2-4 месяцев после рождения). Для пациентов с гидроцефалией операция может также включать введение трубки (шунта) для оттока избыточной спинномозговой жидкости (СМЖ) из головного мозга. СМЖ сливается в другую часть организма, где он абсорбируется.
Корректирующая и реконструктивная хирургия может быть рекомендована для коррекции черепно-лицевых пороков развития. Хирургия также может помочь исправить полидактилию и синдактилию, а также другие скелетные дефекты или физические отклонения. Для больных с врожденными пороками сердца может потребоваться лечение определенными препаратами, хирургическое вмешательство и/или другие терапевтические меры. Для некоторых больных с нарушениями слуха полезны слуховые аппараты.
Раннее вмешательство может быть важным для обеспечения того, чтобы дети с синдромом Аперта полностью раскрыли свой потенциал. Специальные терапевтические мероприятия, такие как физиотерапия, трудотерапия и специализированное обучение также будут полезными.
Для пострадавших лиц и их семей рекомендуется генетическое консультирование. Генетический консультант сможет объяснить причины заболевания. Он также может обсудить возможность рождения новых детей с заболеванием. Дополнительно, для всей семьи необходима психосоциальная поддержка.
Прогноз
Прогноз для детей с синдромом Апера зависит от того, насколько серьезным является состояние и на какие системы организма оно повлияло. Заболевание может быть более серьезным, если повлияет на дыхательную систему ребенка или если внутри черепа вырастит давление, но эти проблемы можно исправить хирургическим путем.
Дети с синдромом часто имеют проблемы с обучением. Некоторые дети страдают более серьезно, чем другие.
Поскольку тяжесть болезни может варьироваться в широких пределах, сложно прогнозировать продолжительность жизни. Заболевание может не оказывать существенного влияния на продолжительность жизни ребенка, особенно если у него нет пороков сердца.
Источник
Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 25 декабря 2015;
проверки требуют 12 правок.
Синдром Апера (Аперта) (акрокраниодисфалангия, акросфеносиндактилия, акроцефалосиндактилия) — врожденная аномалия развития черепа, которая сочетается с отклонением развития кистей рук. Раннее закрытие венечного и стреловидного швов способствует деформации черепа, что приводит к внутричерепной гипертензии[1]. Синдром Апера является одной из форм акроцефалосиндактилии[прим. 1].
Впервые это заболевание описал французский педиатр Эжен Аперт в 1906 году[2]. Заболевание встречается у одного новорожденного на 160 000—200 000[3].
Этиология[править | править код]
Акрокраниодисфалангия — заболевание с аутосомно-доминантным типом наследования[прим. 2]. У детей, родители которых страдают синдромом Аперта, вероятность унаследовать заболевание равна 50%. В 1995 году были опубликованы доказательства, судя по которым, причиной возникновения данного синдрома является нарушение работы фибробластов генов 10-й хромосомы человека[4].
Признаки и симптомы[править | править код]
Синдактилия двух пальцев у младенца
Одним из признаков заболевания является то, что краниосиностоз[прим. 3] сочетается с брахикефалией. В связи с преждевременным срастанием коронарных швов увеличивается внутричерепное давление, что обычно приводит к умственной отсталости. Немаловажными признаками синдрома являются высокий, выпуклый лоб, плоское или вогнутое лицо, в результате чего наблюдается нарушение костей лицевого черепа, что приводит к деформации челюсти, а также синдактилия рук и ног с вовлечением 2, 3 и 4-го пальцев.
Лечение[править | править код]
Радикальных методов лечения не существует.
Симптоматическое лечение данной патологии заключается в хирургическом увеличении объёма черепа, коррекция синдактилии и полидактилии.
Хирургическое лечение включает в себя раннюю краниоэктомию коронарного шва и фронто-орбитальную репозицию для уменьшения проявлений дисморфизма и патологических изменений формы черепа. Операции по поводу синдрома Апера часто состоят из нескольких этапов, последний проводится в подростковом возрасте. Первый этап часто выполняется уже в 3 мес.[5]
Консервативные методы лечения применяют для разработки суставов. Также, для стимуляции психического развития назначают ноотропные препараты: аминалон, пирацетам и другие[1], использовались в эпоху развития науки до проверки лечения методами доказательной медицины.
Примечания[править | править код]
- ↑ Акроцефалосиндактилия — группа наследственных пороков развития черепа и пальцев.
- ↑ Аутосомно-доминантный тип наследования — тип наследования, при котором мутантный аллель доминирует над нормальным аллелем, в связи с чем болезнь или признаки болезни ярко выражены.
- ↑ Краниосиностоз — преждевременное сращение некоторых костей черепа.
Источники[править | править код]
- ↑ 1 2 Л.О. Бадалян. Детская неврология. — Москва: Медицина, 1984. — С. 347—349. — 576 с.
- ↑ Apert’s syndrome (whonamedit.com) (англ.). Дата обращения 13 августа 2010. Архивировано 6 мая 2012 года.
- ↑ Kaplan, L C. Clinical assessment and multispecialty management of Apert syndrome (англ.) // Clinics in plastic surgery : journal. — 1991. — April (vol. 18, no. 2). — P. 217—225. — ISSN 00941298. — PMID 2065483.
- ↑ Wilkie, A O; S. F. Slaney, M. Oldridge, M. D. Poole, G. J. Ashworth, A. D. Hockley, R. D. Hayward, D. J. David, L. J. Pulleyn, P. Rutland. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome (англ.) // Nature genetics : journal. — 1995. — February (vol. 9, no. 2). — P. 165—172. — doi:10.1038/ng0295-165. — PMID 7719344.
- ↑ Синдром Апера: клинические проявления и этиология — Интернет-сообщество нейрохирургов Росcии. neuro-online.ru. Дата обращения 4 марта 2017.
Источник
Синдромные краниосиностозы: синдром Крузона, синдром АпертаПриблизительно 15-20% краниосиностозов входят в состав различных синдромов, большая часть которых генетически детерминирована. Имеется полный обзор синдромных краниосиностозов (Cohen, 1986, 1988) и подробное описание клинической картины и генетических аспектов (Britto и Reardon, 2004; Thompson и Britto, 2004). Некоторые из наиболее часто встречающихся синдромов представлены в таблице 5.2. Такие синдромы представлены множеством аномалий, одной из которых является краниосиностоз. В рамках одного и того же синдрома тип краниосиностоза может варьировать и не являться определяющим тяжесть заболевания фактором; аномалии органов черепа могут иметь большее значение, так как приводят к нарушениям дыхания, питания и зрения. Диагностика не должна ограничиваться исключительно определением пораженных швов. На синдромы Крузона и Апера приходится приблизительно по трети случаев синдромных краниосиностозов, а оставшаяся треть встречается при комплексных или редких синдромах, отчасти пока не классифицированных (Le Merrer et al., 1988, Lajeunie et al., 1995). а) Синдром Крузона. Синдром Крузона характеризуется аутосомно-доминантным типом наследования. Более чем в половине из 61 случая (Kreiborg, 1981) отмечались вновь возникшие мутации; данный синдром объяснял наличие 3% краниосиностозов среди 370 пациентов (Hunter и Rudd, 1977). Установлено, что синдром Крузона и Аперта объясняют 4,8% и 4,5% случаев краниосиностозов при рождении (Cohen и Kreiborg, 1990). Фенотип варьирует даже внутри одной генеалогической линии; часто встречаются слабо выраженные случаи, а внешний вид взрослых может казаться нормальным, но ранние фотографии родителей могут помочь в выявлении минимальных поражений. Ген синдрома Крузона располагается на участке 10q25-q26 (Preston et al., 1994) и кодирует рецептор фактора роста фибробластов-2 (Jabs et al., 1994). Выявлено несколько мутаций данного гена (Reardon et al., 1994; Oldridge et al., 1995). Существенными проявлениями данного синдрома являются гипоплазия верхней челюсти, неглубокие глазницы и проптоз. Краниосиностоз является постоянным проявлением синдрома, развивается на первом году жизни и чаще всего вначале поражает венечные швы. В итоге происходит поражение всех швов, но форма черепа варьирует от трилистника (Rohatgi, 1991) до скафоцефалии. Часто отмечается выступание черепа в области закрытия переднего родничка. Среди пациентов с синдромом Крузона отмечается выраженная вариабельность проявлений. Часто встречается проводниковая тугоухость поэтому необходимо целенаправленное обследование. Постоянно встречающиеся аномалии внутренних органов и конечностей отсутствуют, но обструкция носовой полости и глотки может приводить к хронической дыхательной недостаточности и формированию легочного сердца (Moore, 1993). Припадки встречаются у 12% пациентов (работы Kreiborg), но задержка умственного развития выявляется редко (3%). Гидроцефалия встречается редко (Marchac и Renier, 1982), но повышение внутричерепного давления было выявлено в 37% случаев, также зарегистрированы случаи хронической грыжи миндалин мозжечка (Cinalli et al., 1995). Аномалии костей запястья и пястных костей встречаются часто и напоминают проявления синдрома Пфейффера. Учитывая множественные поражения лица, обструкцию дыхательных путей и верхних отделов желудочно-кишечного тракта, хирургическое лечение при раннем проведении дает удовлетворительные результаты.
б) Синдром Аперта. Синдром Аперта практически всегда наследуется аутосомно-доминантным путем. Заболевание аллельно синдрому Крузона и вызвано мутацией гена рецептора фактора роста фибробластов-2 (Wilkie et al., 1995). Синдром характеризуется краниосиностозом, мальформациями средней линии лица и симметричной синдактилией кистей и стоп с поражением второго, третьего и четвертого пальцев (Cohen, 1986) (рис. 5.6). Обычно отмечается раннее поражение венечного шва, с формированием высокого брахицефалического черепа, но синостоз имеет вариабельный характер. Распространенный открытый дефект свода черепа начинается от корня носа, продолжается до заднего родничка новорожденных и закрывается до трехлетнего возраста с формированием выпячивания черепа. Передняя черепная ямка короткая, а глазницы неглубокие. Внутричерепной объем превышает контрольные показатели. Часто встречается расщелина неба. Задержка умственного развития встречается очень часто в очень тяжелой степени. Тем не менее, по результатам одного из исследований задержка умственного развития отмечалась только у половины пациентов (Patton et al., 1988). Достаточно часто встречается гидроцефалия, а аномалии (особенно частичный агенез мозолистого тела) отмечаются у 12% пациентов (Cohen и Kreiborg, 1990). Сращение пятого и шестого шейных позвонков отмечается в 70% случаев; часто встречается глухота и атрофия зрительного нерва. Приблизительно у 10% пациентов выявляются аномалии внутренних органов. Даже при использовании современных реконструктивных методик прогноз неблагоприятный и может быть еще более серьезным при сочетанных аномалиях, таких как аномалии сердца, сколиоз или микроофтальмия.
в) Другие синдромные краниосиностозы. Следующими по распространенности являются синдром Сетре-Чотзена (de Heer et а1., 2005) и синдром Пфайффера (Vanek и Losan, 1982; Zankl et al., 2004). Прогноз для когнитивных функций обычно благоприятный, но синдромы характеризуются заметной вариабельностью неврологических и морфологических проявлений и иногда сопровождаются тяжелыми деформациями. При синдроме Сетре-Чотзена обычно встречается двусторонний синостоз венечного шва, часто асимметричный (Reardon и Winter, 1994). Аномалии лица, глаз (птоз), внешнего уха и пальцев рук имеют легкую степень выраженности. Синдром имеет преимущественно семейный характер с часто встречающимися малыми формами (Chun et al., 2002). Синдром Пфайффера также является аутосомным доминантным заболеванием и представлен сочетанием краниосиностоза (преимущественно венечного шва) и характерных аномалий конечностей. Синдром включает широкие и короткие большие пальцы стоп и кистей, симфалангию кистей, частичную мягкотканную синдактилию кистей и стоп. Часто встречается проводниковая тугоухость от легкой до умеренной степени выраженности. Интеллект обычно в пределах нормы. Описано три подтипа заболевания различной степени выраженности (Cohen, 1993b). Описано большое количество минимально выраженных синдромных краниосиностозов (Thompson и Britto 2004); в настоящее время выделено не менее 67 синдромов (Cohen, 1986), включающих краниосиностозы, и ожидается продолжение. Генетическое консультирование в таких случаях обычно носит неточный характер, но в 7 из 11 случаев недиагностированных синдромных краниосиностозов была выявлена семейная история заболевания (Le Merrer et al.,. 1988), таким образом, вероятность рождения больных детей достаточно высока.
г) Молекулярные аспекты и аспекты развития синдромных краниосиностозов. Последние несколько лет существенное внимание уделяется генетическим аспектам синдромных краниосиностозов (Superti-Furga et al., 2001). Синдромы Аперта, Крузона и Пфейффера связаны с мутациями гена рецептора фактора роста фибробластов-2. Синдром Пфейффера также может быть связан с мутацией гена рецептора фактора роста фибробластов-1. В целом 30 синдромных краниосиностозов расцениваются как моногеные заболевания (Britto и Reardon, 2004). Несиндромные краниосиностозы, особенно венечный синостоз, также могут быть связаны с мутациями гена рецептора фактора роста фибробластов-1 (Lajeunie et al, 1995). Причины формирования различных фенотипов при мутации в одном и том же гене неясны. Известно большое количество различных мутаций генов рецептора фактора роста фибробластов (Kannu и Aftimos, 2007); существует некоторая корреляция между генотипом и фенотипом, но определенное воздействие дают также эпигенетические факторы и факторы внешней среды. Мутации, являющиеся причиной заболевания, в основном представляют собой миссенс-мутации, а их воздействие основано на «усилении функции»; исключение составляет связанный с синдромом Сетре-Чотзена ген TWIST на 7-й хромосоме, гаплонедостаточность которого служит причинной развития синдрома. д) Лечение краниосиностозов. В течение последнего десятилетия наметился явный прогресс в лечении краниосионостозов. Многие аспекты лечения невозможно обсудить в рамках данной статьи; для получения исчерпывающей информации следует обратиться к монографии по данной проблеме, вышедшей под редакцией Hayward et al. (2004). В случае сагиттального и одностороннего венечного синостоза и во многих случаях двустороннего венечного и сложного синостоза достигаются удовлетворительные косметические результаты лечения. Наилучшие результаты были достигнуты при раннем оперативном лечении в возрасте до 6 месяцев, но значимая коррекция достигалась позже. Описаны различные комплексные методы лечения (Marchac и Renier, 1982; Marsh и Schwartz, 1983), заключающиеся в обширных хирургических вмешательствах. Общепризнанно, что хирургическое лечение показано в случае повышения внутричерепного давления и/или при начальной стадии атрофии зрительного нерва. В некоторых случаях, таких как сагиттальный синостоз или плагиоцефалия, риск неврологических осложнений невысок, а целью вмешательства является только коррекция формы черепа. При использовании современных методик лечения изолированных синостозов риск осложнений не столь высок, поэтому хирургическое лечение показано даже при таких относительно доброкачественных формах как сагиттальный синостоз, так как косметические дефекты могут приводить к значимому смущению и затруднению общения в школьном возрасте или позднее. Тем не менее, в ходе недавнего исследования с участием 30 пациентов было продемонстрировано, что у неоперированных пациентов к концу первого десятилетия (9,25 лет) отмечалось нормальное интеллектуальное и психологическое развитие, таким образом, вопрос о необходимости систематического вмешательства остается нерешенным (Boltshauser et al., 2004). Цель лечения должна быть разъяснена родителям. Синдромные синостозы могут быть причиной серьезных отклонений в связи с поражением органов черепа и множеством дефектов скелета и нервной системы. Проблема решается лишь при участии специалистов, знакомых с такими специфическими задачами. Особое внимание уделяется слуху, речи и языку, питанию и психологическим проблемам (Hayward et al., 2004). Необходимо осознать, что лечение не ограничивается хирургическими методами. Важными являются офтальмологические аспекты. Частота атрофии зрительного нерва снизилась в результате раннего и эффективного хирургического лечения; данное осложнение в 35,5% случаев имело двусторонний и в 9,1% случаев — односторонний характер и было вызвано косоглазием, астигматизмом, гиперметропией или анизометропией; необходима коррекция и наблюдение специалиста (Тау et al., 2006). Часто встречающаяся обструкция дыхательных путей требует консультации ЛОР-специалиста. — Также рекомендуем «Дисплазии черепа с поражением нервной системы» Редактор: Искандер Милевски. Дата публикации: 6.12.2018 |
Источник