Аритмогенная кардиомиопатия мкб 10 код

Содержание
- Описание
- Дополнительные факты
- Причины
- Классификация
- Симптомы
- Диагностика
- Дифференциальная диагностика
- Лечение
- Прогноз
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Аритмогенная правожелудочковая кардиомиопатия.
Аритмогенная правожелудочковая кардиомиопатия
Описание
Аритмогенная правожелудочковая кардиомиопатия. Заболевание предположительно генетической природы, характеризующееся структурными изменениями правого желудочка и развитием аритмии. Варианты течения варьируются от бессимптомных форм до форм с выраженной тахиаритмией, экстрасистолией, кардиалгией и сердечной недостаточностью. Диагностика осуществляется при помощи эхокардиографических, электрокардиографических, магнитно-резонансных исследований, а также биопсии миокарда. Специфическое лечение отсутствует, терапия сводится к устранению аритмии и застойной сердечной недостаточности. При отсутствии эффекта медикаментозной терапии применяют имплантацию кардиовертер-дефибриллятора.
Дополнительные факты
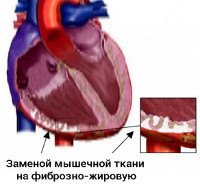
Аритмогенная правожелудочковая кардиомиопатия (аритмогенная дисплазия правого желудочка, аритмогенная болезнь правого желудочка, АП КМП) – заболевание, поражающее стенку правого желудочка, при котором в толще миокарда формируются патологические очаги жировой и фибринозной инфильтрации, иногда с присоединением воспаления (миокардит). Патология была описана под названием «аритмогенная дисплазия правого желудочка» в 1977 году G. Fontaine, после этого исследования продолжил F. I. Marcus, который в 1982 году дал заболеванию современное название.
Встречаемость в различных регионах колеблется в пределах 1-6:10000 жителей; среди обнаруженных больных подавляющее большинство составляют мужчины младше 40 лет, половое распределение — 4:1. Имеет тенденцию к наследственной передаче, поэтому в настоящее время большинство форм АП КМП определяют как аутосомно-доминантное заболевание с неполной пенетрантностью. Интерес к заболеванию резко возрос в связи с выявлением его роли в развитии внезапной сердечной смерти. Так, гистологические исследования миокарда у детей и подростков до 20 лет, причиной смерти которых явилась сердечно-сосудистая патология, показали, что изменения, ассоциированные с АП КМП, были обнаружены в 26% случаев.
Аритмогенная правожелудочковая кардиомиопатия
Причины
В настоящий момент общепризнанной точки зрения на причины развития АП КМП нет ввиду гетерогенности проявлений заболевания. Возможно, АП КМП объединяет в себе несколько схожих по проявлениям патологий с различной этиологией. Но единственной документально подтвержденной теорией на сегодняшний день является наследственная, объясняющая возникновение аритмогенной правожелудочковой кардиомиопатии генетической мутацией.
При изучении генома больных АП КМП были выявлены аномалии генов в 12-й, 14-й, 17-й и 18-й хромосомах – указанные гены кодируют такие белки миокарда как альфа-актин, десмоплакин, плакоглобин, плакофиллин и другие. Нарушения структуры этих белков ведут к понижению устойчивости кардиомиоцитов к повреждающим факторам, что и приводит со временем к жировой инфильтрации. Однако главную роль в развитии аритмии при АП КМП играет нарушение функций белка десмосом, в результате чего распространение возбуждения по миокарду изменяется.
В некоторых случаях вместо очаговой жировой инфильтрации стенок правого желудочка наблюдается фибринозная, имеющая воспалительный характер и в целом напоминающая картину при вирусном миокардите, вызванном вирусом Коксаки и тд Такая форма имеет тенденцию к распространению на левый желудочек и характеризуется тяжелым течением, часто приводящим к смерти больного. С точки зрения наследственной теории развития АП КМП, считается, что мутации генов повышают предрасположенность миокарда к поражению вирусами.
Большинство мутаций наследуются по аутосомно-доминантному типу с пенетрантностью 30-50%. Одна крайне редкая форма аритмогенной правожелудочковой кардиомиопатии (болезнь Наксоса — описано всего 25 случаев) имеет аутосомно-рецессивный характер и высокую пенетрантность – более 90%. Гомозиготы по мутантному гену страдают от злокачественной желудочковой аритмии и часто умирают в детстве или подростковом возрасте.
Классификация
Ввиду выраженной гетерогенности клинических форм АП КМП неоднократно принимались попытки систематизировать и классифицировать виды этого заболевания. В настоящее время выделяют следующие типы аритмогенной правожелудочковой кардиомиопатии:
• Чистая, или эталонная, форма.
• Болезнь Наксоса, характеризующая аутосомно-рецессивным наследованием и злокачественными желудочковыми аритмиями.
• Венецианская кардиомиопатия – нередко распространяется на стенку левого желудочка, имеет выраженный наследственный характер (пенетрантность около 50%), больные могут умереть в детском возрасте.
• Болезнь Покури – форма АП КМП, выявленная в Японии, служит причиной внезапной сердечной смерти подростков.
• Тахикардия, вызванная очагом возбуждения в правом желудочке без экстрасистол или проявлений сердечной недостаточности.
• Редкие желудочковые экстрасистолы, источник которых – очаг возбуждения в стенке правого желудочка, ассоциируемый с участком воспаления. Такая форма АП КМП может осложняться миокардитом с летальным исходом.
• Аномалия Уля – редкая форма АП КМП, характеризующая нарастанием сердечной недостаточности и смертью. При гистологическом исследовании сердца обнаруживается полное замещение кардиомиоцитов жировой и фибринозной тканью.
• Неаритмогенная форма – в большинстве случаев ничем себя не проявляет, именно с ней ассоциируют бессимптомные случаи внезапной сердечной смерти.
Симптомы
Клинические формы АП КМП разделяются на четыре основные группы. При бессимптомной форме патология никак не проявляет себя при жизни больного, в том числе и при электрокардиографических исследованиях. Аритмическая форма характеризуется развитием тахиаритмии, желудочковой экстрасистолии и появлением других электрокардиографических признаков. Субъективные симптомы, как правило, отсутствуют. При развитии выраженной клинической формы на фоне тахиаритмий возникают кардиалгии, приступы сердцебиения, головокружения. Наиболее тяжелым типом клинических проявлений АП КМП является развитие сердечной недостаточности по правожелудочковому типу с характерным симптомокомплексом – венозным застоем, отеками, асцитом. При этом сердечная недостаточность может протекать как на фоне нарушения ритма сердца, так и без него.
Боль в груди слева. Боль в грудной клетке.
Диагностика
Для диагностики АП КМП в кардиологии используется весь спектр современных методик исследования функции сердца. На ЭхоКГ определяется конечный диастолический размер (КДР) и конечный систолический размер (КСР) желудочков, после чего эти данные сравниваются между собой. Если отношение КДР правого желудочка к КДР левого составляет 0,5 и более, это свидетельствует в пользу наличия АП КМП. К ЭКГ-признакам аритмогенной правожелудочковой кардиомиопатии относятся удлинение желудочкового комплекса свыше 110 мс на отведении V1, эпсилон-волна на сегменте ST в отведениях V1-V3, инверсия зубца Т в грудных отведениях из-за замедления деполяризации правого желудочка, а также наличие тахиаритмии и желудочковых экстрасистол.
На рентгеноконтрастной вентрикулографии можно определить дилятацию правого желудочка, причем в ряде случаев прямо визуализируется выпячивание (аневризма) в области очага фибролипозной дисплазии миокарда. В последние годы для специфической диагностики АП КМП применяется магнитно-резонансная томография с гадолиниевым контрастированием, позволяющая не только получить трехмерное изображение сердца, но и дифференцировать жировые и фибринозные очаги от неизмененного миокарда. Для уточнения диагноза практикуют биопсию миокарда с последующим гистологическим изучением тканей – при АП КМП будет наблюдаться жировая инфильтрация, изменение цвета и формы десмосом, а также уменьшение их количества.
Важным признаком является семейный характер симптомов или наличие у больного родственников, скончавшихся от внезапной сердечной смерти или фибрилляции желудочков.
Дифференциальная диагностика
Дифференциальную диагностику производят с идиопатическими формами желудочковых тахиаритмий.
Лечение
Медикаментозное лечение АП КМП включает в себя антиаритмические препараты (амиодарон, соталол). Снижение выраженности тахиаритмий играет важную роль в сохранении жизни пациента, контроль эффективности препаратов кардиолог производит при помощи холтеровского мониторинга. В тех случаях, когда медикаментозная терапия малоэффективна, прибегают к имплантации кардиовертер-дефибриллятора или кардиостимулятора. При развитии сердечной недостаточности используют ингибиторы АПФ, карведилол.
Разрабатываются техники хирургического лечения аритмогенной правожелудочковой кардиомиопатии (вентрикулотомия), которые сводятся к удалению патологических очагов с ушиванием миокарда. Первые результаты таких операций оптимистичны, однако рецидивы возникают в 30-40% случаев. При выраженной сердечной недостаточности эффективным методом лечения будет трансплантация сердца.
Прогноз
Прогноз неопределенный по причине высокой вариабельности проявлений АП КМП. Фибрилляция желудочков с летальным исходом может развиться в любой момент, особенно при отсутствии лечения. При регулярной антиаритмической терапии риск летального исхода снижается примерно на треть. Наилучшие результаты показывает сочетание кардиостимуляторов и медикаментозной терапии, которое снижает риск возникновения летальной фибрилляции практически до нуля. При развитии сердечной недостаточности прогноз, как правило, неблагоприятный.
Основные медуслуги по стандартам лечения | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Клиники для лечения с лучшими ценами
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Источник
Рубрика МКБ-10: I42.8
МКБ-10 / I00-I99 КЛАСС IX Болезни системы кровообращения / I30-I52 Другие болезни сердца / I42 Кардиомиопатия
Определение и общие сведения[править]
Аритмогенная дисплазия правого желудочка
Синонимы: аритмогенная кардиомиопатия правого желудочка
Аритмогенная дисплазия правого желудочка (АДПЖ) — заболевание, при котором нормальный миокард правого желудочка замещается жировой или фиброзно-жировой тканью. Обычно возникает изолированное поражение правого желудочка, однако, в процесс может вовлекаться межжелудочковая перегородка и миокард левого желудочка.
Эпидемиология
Распространённость заболевания в популяции зависит от региона и находится в пределах от 6 до 44 случаев на 10 000 населения. Наибольшую распространенность отмечают в средиземноморских регионах. В 80% случаев ее выявляют в возрасте до 40 лет, чаще у мужчин. Аритмогенная дисплазия правого желудочка выступает причиной 5-20% случаев внезапной смерти в молодом возрасте, занимая второе место после ГКМП.
У более чем половины пациентов аритмогенная кардиомиопатия правого желудочка является семейной, передается главным образом аутосомно-доминантно с переменной пенетрантностью и полиморфной экспрессией. Болезнь Наксоса и синдром Карвахаля показывают аутосомно-рецессивный способ наследования.
Этиология и патогенез[править]
Аритмогенная кардиомиопатия правого желудочка является результатом фиброзно-жировой дисплазии миокарда. Считается, что она является нарушением десмосом с дефектом контакта между клетками и сигналами. Мутации наблюдаются в генах, кодирующих белки сердечных десмосом (JUP, DSP, PKP2, DSG2 и DSC2). Однако у меньшего количества пациентов были обнаружены также мутации в не-десмосомных генах (TGFβ3, TMEM43 / LUMA, DES, CTNNA3, PLN, LMNA, TTN). Обнаружена переменная пенетрантность, предполагающая роль дополнительных генетических или экологических модификаторов. Компаундные / дигенические гетерозиготы идентифицируется у до 25% носителей мутаций и, по-видимому, является дополнительным фактором риска. Недавние исследования указывают на роль канонического сигнального пути Wnt в патогенезе аритмогенной дисплазии правого желудочка.
Макроскопически у больных с АДПЖ выявляют локальную или генерализованную дилатацию ПЖ с истончением миокарда. Типичная локализация изменений: верхушка, инфундибулярная и субтрикуспидальная область («треугольник дисплазии»). Микроскопический критерий диагноза — наличие очагов фиброзно-жировой ткани, перемежающихся с неизменённым миокардом.
Клинические проявления[править]
Клиника аритмогенной кардиомиопатии правого желудочка варьирует от бессимптомных форм до случаев внезапной смерти или тяжелой бивентрикулярной сердечной недостаточности.
Аритмогенная дисплазия правого желудочка обычно дебютирует желудочковыми нарушениями ритма сердца: экстрасистолией различных градаций, короткими «пробежками» желудочковой тахикардии, а в ряде случаев — пароксизмами устойчивой желудочковой тахикардии. Поскольку аритмогенный очаг находится в ПЖ, эктопические желудочковые комплексы имеют вид блокады левой ножки пучка Гиса.
Могут возникать нетипичные боли в груди, слабость, повышенная утомляемость, эпизоды учащённого сердцебиения при физической нагрузке. Аритмогенные коллапсы могут возникать во время нагрузки или спонтанно.
Аритмогенная дисплазия правого желудочка может сопровождаться ладонно-подошвенной кератодемией и шерстистыми волосами, являясь частью фенотипа сердечно-кожных синдромов — болезни Наксоса и синдрома Карвахаля.
Другие кардиомиопатии: Диагностика[править]
Анамнез
Анамнестические сведения, как правило, малоинформативны для постановки диагноза. Следует учитывать возможность семейного характера заболевания, более высокую частоту АДПЖ у жителей средиземноморского региона.
Физикальное исследование
При физикальном исследовании в половине случаев не выявляют никаких отклонений.
Лабораторные исследования
Для диагностики АДПЖ лабораторные исследования диагностической ценности не имеют. Тем не менее, стандартные клинические и биохимические исследования должны входить в программу обследования больных с установленным или предполагаемым диагнозом АДПЖ.
Инструментальные исследования
1. Электрофизиологическое исследование сердца. Его выполняют для уточнения характера нарушений ритма и оценки риска фатальных аритмий.
2. Методы визуализации имеют большое значение для диагностики АДПЖ. При вентрикулографии и ЭхоКГ (в том числе контрастной) выявляют аномалии сократимости ПЖ. При выполнении МРТ выявляют повышенное содержание жировой ткани в миокарде. В ряде случаев эта методика может заменить ангиографию и, возможно, биопсию в диагностике этой патологии.
3. Эндомиокардиальная биопсия. С её помощью можно выявить достоверные диагностические признаки АДПЖ. Биопсию выполняют в области межжелудочковой перегородки и свободной стенки ПЖ. Чувствительность метода составляет около 20%, так как не всегда удаётся взять биопсию именно из поражённого участка.
Дифференциальный диагноз[править]
На поздних стадиях у больных может возникнуть недостаточность кровообращения, что вызывает серьёзные трудности при дифференциальной диагностике АДПЖ с ДКМП (Дилатационная кардиомиопатия).
Другие кардиомиопатии: Лечение[править]
Цели лечения
Заболевание имеет неуклонно прогрессирующий характер, однако, при своевременном выявлении и адекватном лечении можно существенно улучшить прогноз. Терапия при АДПЖ направлена на предупреждение ВСС и лечение сердечной недостаточности.
Показания к госпитализации
Пациентов с подозрением на АДПЖ для уточнения диагноза госпитализируют в специализированные кардиологические и кардиохирургические учреждения. При установленном диагнозе АДПЖ повторные госпитализации необходимы при рецидивирующих нарушениях ритма, а также при декомпенсации сердечной недостаточности.
Немедикаментозное лечение
Пациентам с признаками сердечной недостаточности необходимо ограничить потребление поваренной соли и жидкости. Всем пациентам с АДПЖ необходимо исключить интенсивные физические нагрузки.
Медикаментозное лечение
Лечение ХСН при АДПЖ предполагает назначение стандартной терапии: диуретиков, ингибиторов АПФ, дигоксина, при наличии показаний используют антикоагулянты.
Среди антиаритмиков наибольший опыт накоплен в отношении амиодарона и соталола. Последний имеет наибольшую эффективность, поэтому с целью лечения желудочковых аритмий и профилактики ВСС рекомендуют назначать, в первую очередь, соталола.
Хирургическое лечение
При неэффективности антиаритмической терапии следует использовать хирургические методы, в частности, имплантацию кардиовертера-дефибриллятора. Проведение радиочастотной абляции имеет низкую эффективность, так как развиваются рецидивы аритмий, вызванные активизацией новых очагов. Пациентам, рефрактерным к консервативной терапии, показана трансплантация сердца.
Информация для пациентов
Пациенту необходимо разъяснить характер заболевания, дать рекомендации по диете, потреблению жидкости, режиму физической активности. Важно предупреждать больных о возможности прогрессирования заболевания, а также информировать о симптомах, требующих обращения за медицинской помощью. Необходимо профилактическое обследование родственников пациента для исключения семейных форм АДПЖ.
Прогноз
АДПЖ — прогрессирующе заболевание, и при отсутствии лечения высока вероятность ВСС (внезапная сердечная смерть). Данные о долгосрочном прогнозе АДПЖ отсутствуют. Пятилетняя летальность, по разным данным, составляет от 4 до 19%.
Профилактика[править]
Не разработана.
Прочее[править]
Кардиомиопатия такоцубо
Синонимы: синдром тако-цубо, ампуллярная кардиомиопатия, апикальный баллонный синдром,
баллонная кардиомиопатия, синдром разбитого сердца, синдром такоцубо, транзиторный апикальный баллонный синдром левого желудочка, стрессовая кардиомиопатия, TAKO-TSUBO
Определение и общие сведения
Кардиомиопатия такоцубо представляет собой недавно описанный острый сердечный синдром, который имитирует острый инфаркт миокарда и характеризуется ишемическими симптомами, повышением сегмента ST на электрокардиограмме и повышенным уровнем сердечных маркеров.
Синдром такоцубо был первоначально был описан в японской популяции, но недавно был зарегистрирован в США и Европе. Кардиомиопатия такоцубо чаще всего встречается у женщин в постменопаузе в возрасте от 55 до 75 лет, с оценкой заболеваемости в общей популяции 1/36 000.
Этиология и патогенез
Хотя точная причина остается неизвестной, синдром обычно провоцируется острым эмоциональным или физическим стрессом.
Клинические проявления
Пациенты, обычно обнаруживают ишемическую боль в грудной клетке, одышку, повышение сегмента ST и продление интервала QT на ЭКГ, незначительное повышение уровней сердечных ферментов и биомаркеров, а также транзиторное «шаровидное (баллонное)» расширение верхушки и среднего отдела левого желудочка. В отличие от острых коронарных артериальных синдромов, у пациентов с кардиомиопатией такоцубо не наблюдается ангиографически подтвержденной обструкции коронарных артерий. Наиболее распространенными осложнениями синдрома такоцубо являются кардиогенный шок и затруднение оттока из левого желудочка, инсульт и образование апикального тромба.
Диагностика
Диагноз подтверждается коронарной артериографией, вентрикулографией и эхокардиографией левого желудочка.
Дифференциальный диагноз
Дифференциальный диагноз включает острый коронарный артериальный синдром и другие типы транзиторной желудочковой дисфункции.
Лечение
Пациентов следует контролировать и шоспитализировать при предсердных и желудочковых аритмиях, сердечной недостаточности и механических осложнениях. Ведение состоит из поддерживающей и симптоматической терапии бета-блокаторами, ингибиторами АПФ, ацетилсалицилвой кислотой и диуретиками.
Прогноз
Краткосрочный прогноз благоприятный, характеризуется клиническим выздоровлением в течение нескольких недель, когда принимаются соответствующие меры во период острой фазы заболевания. Течение синдрома такоцубо может быть осложнено разрывом левого желудочка, что делает синдром одной из причин внезапной смерти.
Источники (ссылки)[править]
Кардиология [Электронный ресурс] / под ред. Ю. Н. Беленкова, Р. Г. Оганова — М. : ГЭОТАР-Медиа, 2011. — https://www.rosmedlib.ru/book/ISBN9785970427675.html
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
Источник