Аномалия пьера робена в составе синдрома стиклера

Синдром Робéна (аномалия Робена) — врождённый порок челюстно-лицевой области, характеризующийся тремя основными клиническими признаками: недоразвитием нижней челюсти (микрогенией), глоссоптозом (недоразвитием и западанием языка) и наличием расщелины нёба. Встречается на каждые 10—30 тыс. новорожденных.
Описан французским стоматологом Пьером Робеном (фр. Pierre Robin) (1867—1950)
Этиология и патогенез[править | править код]
Факторы возникновения и механизм развития не выяснены. Имеются указания на то, что в основе Робена синдрома лежат нарушения эмбрионального развития, вызванные разнообразной патологией дородового периода.
Клиническая картина и диагностика[править | править код]
Сразу же после рождения появляется резкое нарушение дыхания, связанное с западением языка. Ребёнок беспокоен, выражена синюшность, на вдохе ребёнок хрипит с втяжениями грудной клетки. Из-за нарушения акта глотания во время кормления ребёнка может наступить удушье. Характерен внешний вид новорожденного — так называемое «птичье лицо», возникающее в связи с недоразвитием нижней челюсти (от слабо выраженного недоразвития до полного её отсутствия).
Наряду с типичной триадой (нижняя микрогнатия, глоссоптоз и расщелина нёба) у больных с синдромом Робена имеются и другие пороки развития (врождённая катаракта, миопия, пороки сердца, мочеполовой системы, аномалии развития грудины и позвоночника, а также полидактилия (присутствие лишних пальцев) и врождённое отсутствие конечностей). Умственное недоразвитие отмечается примерно у 20 % больных.
Диагноз основывается на клинических проявлениях этого синдрома.
Лечение и профилактика[править | править код]
Лечение должно проводиться практически сразу после рождения. В лёгких случаях для профилактики нарушения дыхания достаточно держать новорожденного в положении лёжа на животе или вертикально, наклонив голову к груди. При кормлении нельзя держать ребёнка горизонтально из-за опасности попадания пищи в дыхательные пути и развития аспирационной пневмонии с последующим смертельным исходом.
При резко выраженной нижней микрогнатии (недоразвитие нижней челюсти и значительном смещении языка кзади) требуется выведение языка вперёд в физиологическое положение оперативным путём. В случаях средней степени тяжести подтягивают язык вперед и фиксируют его к нижней губе. В тяжелых случаях применяют трахеостомию или компрессионно-дистракционный остеосинтез.[1]
Компрессионно-дистракционный остеосинтез[править | править код]
Лечение у новорожденных и младенцев проводится по жизненным показаниям.
На нижнюю челюсть с 2-х сторон устанавливаются аппараты, проводятся распилы челюсти между лапками аппаратов, и фрагменты челюсти плотно прижимаются друг к другу. Начиная с 5-6 суток после операции, фрагменты челюсти постепенно, со скоростью 1 миллиметр в сутки, разводятся аппаратами на необходимую величину. Между фрагментами кости образуется новая молодая кость — костная мозоль.
Самостоятельное дыхание становится возможным в среднем уже на 4-6 сутки дистракции, которая продолжается до достижения правильного соотношения челюстей. На самостоятельное питание ребёнок переводится, как правило, к моменту окончания дистракции или немного позже. Ребёнок выписывается с аппаратами на время (3 месяца) превращения костной мозоли в полноценную кость. Через 3 месяца — повторная госпитализация для удаления аппаратов.[2]
Расщелина нёба[править | править код]
В более старшем возрасте (1—1,5 года[2]) устраняют расщелину нёба. Показаны занятия с логопедом для установления правильной речи.
Существует альтернативный безоперационный метод, заключающийся в установке на верхнюю челюсть индивидуально изготовленной пластиковой платы. Плата закрывает расщелину нёба и давит на корень языка, выдвигая его вперед[3].
Профилактика[править | править код]
Профилактика заключается в исключении всех неблагоприятных факторов в дородовом периоде развития ребёнка.
Примечания[править | править код]
Ссылки[править | править код]
- Лечение пациентов с синдромом Пьера Робена
Источник
Секвенция (синдром, последовательность, аномалад) Пьера Робена (СПР) — это симптомокомплекс, включающий триаду признаков: микроретрогнатию, неполную расщелину неба или готическое (арковидное, дугообразное) небо, глоссоптоз [1–12].
Французский стоматолог Пьер Робен (1867–1950) в 1923 году на основании 30-летнего наблюдения определил последовательность проявления признаков, которые приводили к обструкции верхних дыхательных путей [2, 3, 5, 7, 9, 12].
Различают СПР изолированную и в составе более 300 синдромов. Изолированная СПР может быть результатом дизрупции/деформации в эмбриогенезе [1–12].
Клиническая картина СПР многообразна, и осложнения различны в разные возрастные периоды жизни ребенка. Ведущим фенотипическим признаком является микрогнатия. Степень выраженности симптомов вариабельна — от незначительной гипоплазии нижней челюсти с арковидным небом, при которой функции жизнеобеспечения не нарушены, дефект носит косметический характер и при своевременной реабилитации не влечет ортодонтических проблем. Тяжелая степень выраженности симптомов ведет к нарушению витальных функций сразу после рождения, в первую очередь к дислокационной асфиксии.
Примерно в 50 % случаев СПР встречаются различные виды расщелин неба: скрытая расщелина неба, расщелина язычка и неполная расщелина. В 10–15 % случаев СПР глоссоптоз сочетается с анкилоглоссией [1–12].
К внечелюстным проявлениям СПР относятся: аномалии ушной раковины и внутреннего уха, глаз, врожденные пороки сердца, патология опорно-двигательного аппарата, мочеполовые мальформации, умственная отсталость [1–12].
В 1965 году американский педиатр Gunnar B. Stickler и соавторы опубликовали результаты длительного наблюдения пациентов с мальформациями орофациальной области, глаз, костно-суставной системы, слуха. Данная патология была определена как прогрессирующая артроофтальмопатия и названа именем автора.
Синдром Стиклера (СС) — аутосомно-доминантная наследственная коллагенопатия (II и IX типы коллагена), имеющая прогрессивное течение и разно-образные проявления в различные возрастные периоды. Популяционная частота 1 : 7500–15 000, соотношение полов (М : Ж) 0,9.
Клинические проявления СС широко варьируют, однако общими симптомами СС являются: орофациальные мальформации (плоское лицо, гипоплазия нижней челюсти, глоссоптоз, арковидное небо и/или различные расщелины неба), глазная патология (миопия, отслойка сетчатки, катаракта, глаукома, аномалия стекловидного тела), дегенеративные заболевания суставов, поражение слуха. Телосложение у пациентов астеническое, грудина воронкообразная, кифоз, сколиоз, вальгусная деформация голеней. Также наблюдаются тугоухость или полная глухота в связи с изменениями во внутреннем ухе.
Манифестация клинических признаков различна и зависит от возраста пациента. Даже в одной семье отмечается значительная вариабельность фенотипов. Развернутая клиническая картина наблюдается после 30-летнего возраста и постоянно прогрессирует. Из общих проявлений синдрома следует отметить увеличение и гиперподвижность суставов у новорожденных и в раннем детстве, в подростковом периоде появляются боль и тугоподвижность в суставах. В юношеском возрасте — прогрессирующий остеоартрит с периодическими обострениями. После 30 лет наблюдается выраженная дегенеративная артропатия тазобедренных, коленных и голеностопных суставов [6, 8–12].
Представляем собственное наблюдение случая СС. Шестимесячный мальчик, рожденный молодыми родителями, без профессиональных и других вредностей, акушерский и семейный анамнезы не отягощены, от первой, нормально протекающей беременности, первых родов, антропометрические показатели соответствовали сроку гестации. Пренатально в сроке 19 недель 5 дней выявлена микрогения, расщелина неба пренатально не диагностирована. Состояние при рождении ребенка тяжелое, нуждался во вспомогательной вентиляции, улучшение состояния отмечалось в положении на животе. У ребенка выявлены микрогнатия, глоссоптоз, расщелина неба, диагностирован синдром Пьера Робена. В периоде новорожденности у ребенка отмечались кислородозависимость, трудности со вскармливанием, патологическая потеря массы тела. После нормализации состояния с рекомендациями по уходу ребенок выписан домой под наблюдение участкового педиатра.
При осмотре в клинике челюстно-лицевой хирургии обращают на себя внимание аномалии краниофациальной области: плоское лицо, микростомия, микроретрогнатия, расщелина твердого и мягкого неба до средней трети. Движения нижней челюсти ограничены. Осмотр ротовой полости затруднен, преддверие уменьшено, неполная расщелина неба до средней трети, дно ротовой полости недоразвито, глоссоптоз (рис. 1). Ребенок обследован: пороки черепно-лицевой области носят изолированный характер. Предварительный диагноз: синдром Пьера Робена.
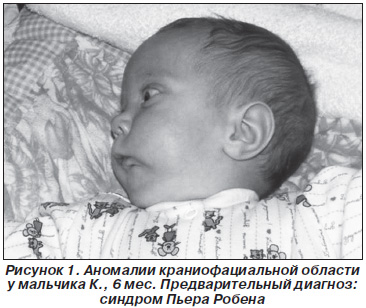
При осмотре у матери ребенка выявлены похожие, но значительно менее выраженные орофациальные проявления: микроретрогнатия, гипоплазия средней трети лица, аномалии зубов, расщелина язычка (рис. 2), при дообследовании диагностирована глазная патология (миопия высокой степени, астигматизм) и суставная патология (незначительные изменения эпифизов локтевых костей).
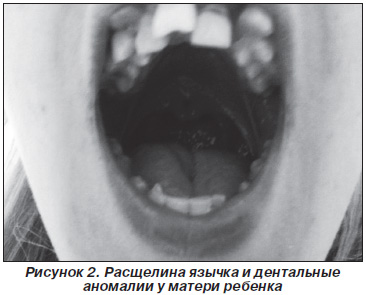
Из анамнеза: мать также родилась преждевременно. Отмечались трудности со вскармливанием и дефицит массы тела на первом году жизни. С 5-летнего возраста на учете у офтальмолога по поводу близорукости. Кариотип матери 46,ХХ, ребенка 46,ХY.
Дифференциально-диагностический ряд представлен фенотипически подобными синдромами — синдром Марфана, Элерса — Данло, но ключевые признаки данных синдромов отсутствовали. После комплексного обследования матери и ребенка диагностирован синдром Стиклера. При сборе семейного анамнеза не отмечены другие родственники, имеющие признаки артроофтальмопатий. У матери пробанда тип наследования определен как аутосомно-доминантный, мутация de novo.
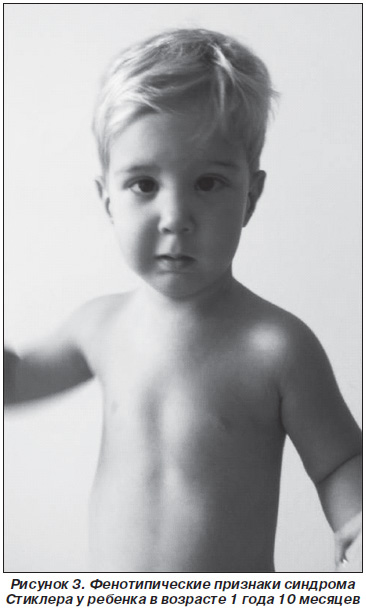
На момент написания статьи мальчику 1 год 10 месяцев. В возрасте 13 месяцев произведена ураностафилопластика. При осмотре у ребенка выявлены: высокое физическое развитие, деформации грудной клетки и конечностей, миопия, астигматизм, страбизм (рис. 3). Однако такой патогномоничный признак, как микрогнатия, регрессирует в результате проводимой комплексной терапии.
Лечение детей с синдромом Пьера Робена поэтапное и длительное, сочетающее консервативные (позиционная терапия, коррекция кормления, терапия сопутствующей патологии) и хирургические методы (трахеостомия, ураностафилопластика и компрессионно-дистракционный остеосинтез). Глоссопексия в настоящее время не применяется. К неотложным мероприятиям относятся устранение обструкции верхних дыхательных путей и коррекция кормления. Ураностафилопластику проводят в возрасте 6–24 месяцев в зависимости от общего состояния пациента. В течение всей жизни, и особенно в раннем возрасте, пациенты должны регулярно наблюдаться у отоларинголога, офтальмолога, ортопеда, логопеда, ортодонта и иммунолога [1, 4, 7–9, 12].
Таким образом, к ранним признакам СС, диагностируемым в периоде новорожденности, относятся лицевые изменения, характерные для СПР, при выявлении которых необходимо дообследование с целью своевременной диагностики и выбора тактики лечения и ведения пациентов. Расщелина неба и более ранние проявления патогномоничных признаков СС можно расценивать как феномен антиципации в данной семье.
Список литературы
1. Хирургическое лечение новорожденных и грудных детей с синдромом Пьера-Робена / Дубин С.А., Комелягин Д.Ю., Злыгарева Н.В. [и др.] // Российский вестник детской хирургии, анестезиологии и реаниматологии. — 2011. — № 2. — С. 33-39.
2. Синдром Пьера Робена у детей / Кириллова Л.Г., Ткачук Л.И., Шевченко А.А. [и др.] // Междунар. неврол. журн. — 2010. — № 3. — С. 18-23.
3. Последовательность Пьера Робена в детской практике / Кириллова Л.Г., Ткачук Л.И., Шевченко А.А. [и др.] // Перинатология и педиатрия. — 2010. — № 2. — С. 32-39.
4. Синдроми вроджених вад, що впливають на зовнішній вигляд обличчя (синдром П’єра-Робена аномалад). МКХ-10 Q 87.0: Протокол надання стоматологічної допомоги // Стоматолог Инфо-Х. — 2010. — № 12. — C. 67.
5. Синдром Пьера Робена в детской практике. Современный взгляд на проблему / О.А. Шевченко // Журнал практичного лікаря. — 2009. — № 1. — С. 29-32.
6. Синдром Стиклера I типа у детей / Семячкина А.Н., Поляков А.В., Новиков П.В. [и др.] // Российский вестник перинатологии и педиатрии. — 2009. — Т. 54. № 3. — С. 45-51.
7. Тактика ведения новорожденных с синдромом Пьера-Робена / Мельникова Е.В., Карачунский М.Г., Тамазян Г.В. [и др.] // Вопросы практической педиатрии. — 2008. — Т. 3. № 5. — С. 36-37.
8. Importance of early diagnosis of Stickler syndrome in newborns / Antunes R.B., Alonso N., Paula R.G. // J. Plast. Reconstr. Aesthet. Surg. — 2012. — Vol. 65. — № 8. — P. 1029-1034.
9. Robin Sequence: From Diagnosis to Development of an Effective Management Plan / Evans K.N., Sie K.C., Hopper R.A. [at al.] // Pediatrics. — 2011. — Vol. 127. — № 5. — P. 936-948.
10. Stickler syndrome, ocular-only variants and a key diagnostic role for the ophthalmologist / Snead M.P., McNinch A.M., Poulson A.V. [at al.] // J. Eye. — 2011. — Vol. 25. — № 11. — P. 1389-1400.
11. Couchouron T. Early-onset progressive osteoarthritis with hereditary progressive ophtalmopathy or Stickler syndrome / T. Couchouron, C. Masson // Joint Bone Spine. — 2011. — Vol. 78. — № 1. — P. 45-49.
12. Lansford M. Focus on the physical assessment of the infant with Stickler syndrome / M. Lansford // Adv. Neonatal Care. — 2008. — Vol. 8. — № 6. — P. 308-314.
Источник
Синдром Робена (аномалия Робена) — врождённый порок челюстно-лицевой области, характеризующийся тремя основными клиническими признаками: недоразвитием нижней челюсти (микрогенией), глоссоптозом (недоразвитием и западанием языка) и наличием расщелины нёба. Встречается на каждые 10—30 тыс. новорожденных.
Описан французским стоматологом Пьером Робеном.
У больных детей нарушаются жизненно важные функции организма — дыхание и глотание.Во время вдоха грудная клетка втягивается, малыши хрипят, а иногда вообще перестают дышать. В роддоме больным интубируют трахею для обеспечения дыхания. Синдром обструктивного апноэ — жизнеугрожающее состояние, нередко приводящее к внезапной смерти. Остановка дыхания обычно возникает в положении лежа на спине. При нарушении глотания питание осуществляться через желудочный зонд. Кормление обычным способом повышает риск попадания воды и пищи в дыхательные пути. Это может привести к развитию пневмонии или острого бронхита, которые угрожают жизни ребенка.
Причины
Рассматривается возможность аутосомно-рецессивного наследования болезни. Различают два вида синдрома в зависимости от этиологии: изолированный и генетически детерминированный. Изолированный вид развивается из-за компрессии нижней части челюсти во период эмбрионального развития. Компрессия может развиваться вследствие:
- Наличие в матке локальных уплотнений (кисты, рубцы, опухоли).
- Многоплодная беременность.

Также развитие челюсти у плода может нарушаться при:
- Вирусных инфекциях, которые перенесла будущая мать во время беременности.
- Нейротрофических нарушениях.
- Недостаточном количестве фолиевой кислоты в организме беременной женщины.
Клинические признаки
Основным признаком болезни является гипоплазия нижней челюсти – её недоразвитость (такие дети имеют скошенный недоразвитый подбородок), в связи с чем уменьшена полость рта и происходит смещение языка назад. На нёбных пластинках малыша формируется щель. Дыхательная и глотательная функция затруднена. Очень часто, при синдроме Пьера Робена, проявляются иные патологии:
- Нарушения строения слухового аппарата, повлекшие за собой потерю слуха.
- Заболевания центральной нервной системы (нарушение двигательной функции, микро- и гидроцефалия, замедление развития речи и психики, припадки эпилепсии).
- Врожденные аномалии зрительной функции – катаракта, близорукость.
- Порок сердца.
- Наличие дополнительных пальцев на конечностях. Отсутствие некоторых конечностей.
- Неправильно сформированная мочеполовая система.
- Нарушение формирования позвоночника и грудной клетки.
- Дети с синдромом Пьера Робена в 20% случаев рождаются с умственной отсталостью.

У ребенка, родившегося с названной патологией, зачастую дыхание хриплое и затрудненное, слизистые и кожа синеют. Если вовремя не оказать помощь, может случиться летальный исход.
Диагностика болезни
Диагностика синдрома не представляет особой сложности, поскольку все симптомы достаточно явные и хорошо выражены. При первичном осмотре врач оценивает общее состояние ребенка и анализирует клиническую картину. Это и является основанием для постановки диагноза. При этом важно пройти консультацию генетика, чтобы исключить наличие других патологий. Диагностикой занимается педиатр-неонатолог, дополнительно требуется консультация ортодонта и генетика.
Лечение
К лечению заболевания необходимо приступать сразу же после его обнаружения. На легкой стадии болезни нужно постоянно держать ребенка вертикально либо в положении лежа на животе. Голову младенца при этом нужно наклонять к груди. Врачи советуют не держать ребенка в горизонтальном положении во время кормления – в противном случае пища будет попадать в дыхательные пути.
Также лечение синдрома Пьера Робена может быть консервативным и оперативным.
При консервативном лечении назначают такие препараты, как:
- «Фенобарбитал»;
- «Клоназепам»;
- «Сибазон»;
- «Кортексин лиофилизат».
Оперативное лечение применяется в первую очередь при смещении корня языка по направлению назад. При симптоме Пьера Робена применяются следующие методы:
- метод Дугласа, основанный на поддержании языка с помощью серебряной нити, которая проводится через нижнюю часть десны и нижнюю губу;
- метод Духамеля, при котором толстую серебряную нить проводят через основание языка пациента и две щеки;
- методы, предполагающие использование ортопедических аппаратов для вытягивания и фиксации языка.
В возрасте одного года допустима операция по устранению расщелины на небе.
Последствия заболевания
Как уже говорилось, дети, перенесшие синдром Пьера Робена, могут немного отставать в развитии интеллекта и речи, у них возможны проблемы со зрением и слухом, а также есть вероятность заболеваний центральной нервной системы. Но проведенное в полном объеме и вовремя начатое лечение способно улучшить не только внешний вид, но и жизнедеятельность ребенка
Загрузка…
Источник
Медицинский эксперт статьи

х
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Синдром Пьера Робена, также известный в медицине под названием аномалия Робена, является врожденной патологией развития челюстной части лица. Свое название заболевание получило в честь французского стоматолога П. Робена, который впервые описал все его признаки. Lannelongue и Menard впервые описали синдром Пьера Робина в 1891 г. в своем докладе на примере 2-х пациентов с микрогнатией, расщелиной нёба и ретроглоссоптозом. В 1926 году Пьер-Робин опубликовал случай заболеввания у младенца с признаками классического синдромом. До 1974 года триада признаков была известна как синдром Робена-Пьера. Тем не менее, этот синдром сейчас используется для описания пороков формообразования при одновременном наличии множественных аномалий.
Код по МКБ-10
Q87.0 Синдромы врожденных аномалий, влияющие преимущественно на внешний вид лица
Эпидемиология
Это гетерогенный врожденный дефект, который имеет распространенность 1 на 8500 живорожденных. Соотношение мужчин к женщинам составляет 1:1, за исключением Х-хромосомой формы.
Среди этих больных, у 50% младенцев расщелина на мягком небе является неполной, остальные рождаются с дугообразным и необычайно высоким небом, но без расщелины.
[1], [2], [3], [4], [5], [6], [7], [8], [9]
Причины Синдрома Пьера Робена
Рассматривается возможность аутосомно-рецессивного наследования болезни. Различают два вида синдрома в зависимости от этиологии: изолированный и генетически детерминированный. Изолированный вид развивается из-за компрессии нижней части челюсти во период эмбрионального развития. Компрессия может развиваться вследствие:
- Наличие в матке локальных уплотнений (кисты, рубцы, опухоли).
- Многоплодная беременность.
Также развитие челюсти у плода может нарушаться при:
- Вирусных инфекциях, которые перенесла будущая мать во время беременности.
- Нейротрофических нарушениях.
- Недостаточном количестве фолиевой кислоты в организме беременной женщины.
[10], [11], [12], [13], [14]
Патогенез
Синдром Пьера Робена проявляется из-за эмбриональных нарушений, которые вызываются самыми разнообразными патологиями в дородовом периоде.
Существует три патофизиологические теории, которые могут объяснить возникновение синдрома Пьера Робена.
Механическая теория: Эта теория наиболее вероятная. Недоразвитие нижнечелюстного аппарата происходит между 7-й и 11-й неделями беременности. Высокое стояние языка в ротовой полости приводит к образования расщелин в небе, из-за этого не происходит закрытие небных пластинок. Эта теория объясняет классическую перевернутую U-образную расселину и отсутствие связанной с ней заячьей губы. В этиологии определенную роль может играть олигогидрамнион, так как отсутствие амниотической жидкости может привести к деформации подбородка и последующего сдавление языка между небных пластинок.
Неврологическое теория: Задержка в неврологическом развитии была отмечена при проведении электромиографии мышц язычка и глоточных столбов, и вкуса из-за задержки проводимости в подъязычном нерве.
Теория диснейрорегуляции ромбовидного мозга: Эта теория основана на нарушении развития ромбовидного мозга в процессе онтогенеза.
Недостаточное развитие нижней части челюсти ребенка приводит к тому, что ротовая полость значительно уменьшается. Это, в свою очередь, вызывает так называемую псевдомакроглоссию, то есть язык смещается к задней части стенки глотки. Такая патология приводит к развитию обструкции дыхательного пути.
Пока младенец плачет или двигается, проходимость дыхательного пути остается нормальной, но как только он засыпает, снова возникает обструкция.
Из-за респираторных нарушений процесс кормления младенца сильно затруднен. В это время практически всегда возникает обструкция дыхательного пути. Если не применять лечебную коррекцию, то такая патология может привести к сильному истощению всего организма и даже к летальному исходу.
[15], [16], [17], [18], [19], [20]
Симптомы Синдрома Пьера Робена
Заболевание отличается трема основными признаками:
- Нижняя микрогнатия (недостаточное развитие нижней части челюсти, встречается в 91,7% случаев заболевания). Она характеризуется втягиванием нижней зубной дуги на 10-12 мм позади верхней арки. Нижняя челюсть имеет небольшое тело, тупой угол. Ребенок достигает нормального развития приблизительно в возрасте 5-6 лет.
- Глоссоптоз (западание языка по причине его недостаточного развития, отмечается в 70-85% случаев).
- Макроглоссия и анкилоглоссия относительно редкие признаки, отмечаются в 10-15% случаев.
- На небе появляется расщелина.
- Брадипноэ и диспноэ.
- Легкий цианоз.
- Асфиксия, которая чаще всего проявляется во время попыток покормить младенца.
- Глотание невозможно или сильно затруднено.
- Позывы к рвоте.
- Аурикулярные аномалии в 75% случаев.
- Потеря слуха проводящего характера встречается у 60% больных, в то время как атрезия наружного слухового канала встречается только у 5% пациентов, недостаточная пневматизация сосцевидного полости височной кости.
- Аномалии внутреннего уха (аплазия боковых полукруглых каналов, большого вестибулярного акведука, потеря волосковых клеток улитки).
- Носовые пороки развития являются нечастыми и представлены в основном из аномалий корня носа.
- Стоматологические пороки развития встречаются в 30% случаев. Ларингомаляция и небно-глоточная недостаточность наблюдаются приблизительно у 10-15% пациентов с синдромом Пьера Робина.
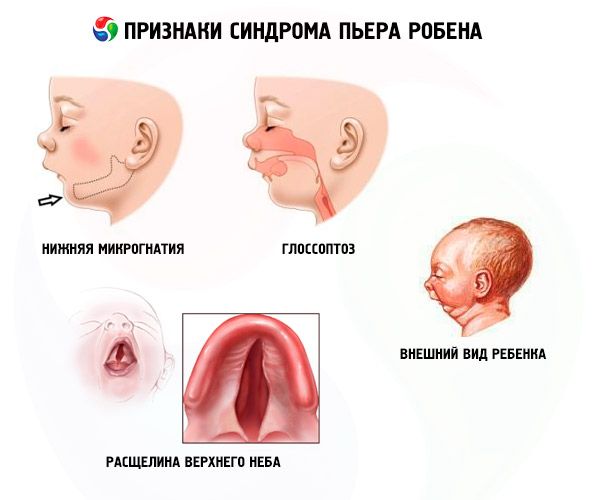
Системные признаки синдрома Пьера Робина
Системные аномалии развития описаны в 10-85% зарегистрированных случаев.
Глазные аномалии встречаются в 10-30% пациентов. Могут встречаться: дальнозоркость, близорукость, астигматизм, склероз роговицы и стеноз носослезного протока.
Сердечно-сосудистые патологии: доброкачественные сердечные шумы, стеноз легочной артерии, открытый артериальный проток, овальное окно, дефект межпредсердной перегородки и легочная гипертензия. Их распространенность варьирует в от 5-58%.
Аномалии, связанные с опорно-двигательным аппаратом (70-80% случаев): синдактилия, диспластические фаланги, полидактилия, клинодактилия, гиперподвижность суставов и олигодактилия верхних конечностей. Аномалии развития нижних конечностей: аномалии стоп (косолапость, аддукция плюсны), бедренные пороки развития (вальгусный или варусный таз, короткие бедра), аномалии бедра (врожденный вывих, контрактуры), аномалии коленного сустава (GENU VALGUS, синхондроз). Пороки развития позвоночного столба: сколиоз, кифоз, лордоз, позвоночная дисплазии, агенезия крестца и копчиковой пазухи.
Патология центральной нервной системы: эпилепсия, задержки развития нервной системы, гидроцефалия. Частота дефектов ЦНС составляет около 50%.
Мочеполовые аномалии: не опустившиеся семенники (25%), гидронефроз (15%), а также водянка яичка (10%).
Ассоциированные синдромы и состояния: синдром Стиклера, синдром трисомии 11q, трисомии 18, синдром удаления 4q, ревматоидная артропатия, гипохондроплазия, синдром Мебиуса.
Стадии
Существует три стадии тяжести заболевания, которые зависят от состояния дыхательных путей ребенка:
- Легкая – есть небольшие проблемы с кормлением, но дыхание почти не затруднено. Лечение проводят в амбулаторно.
- Средняя – дыхание умеренно затруднено, кормление ребенка умеренно трудное. Лечение проводят в стационаре.
- Тяжелая – дыхание очень затруднено, ребенка невозможно нормально кормить. Необходимо использовать специальные приспособления (интраназальный зонд).
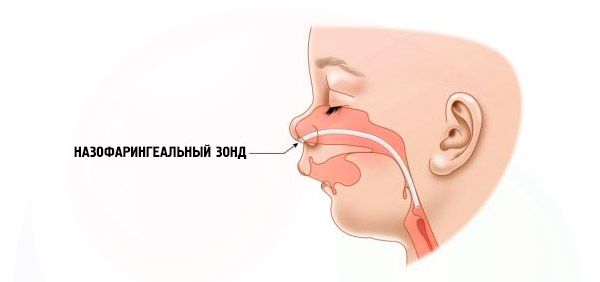
[21], [22], [23], [24]
Осложнения и последствия
Сочетание микрогнатии и глоссоптоза может привести к тяжелым респираторным осложнениям и проблемам во время кормления ребенка.
Синдром Пьера Робена вызывает следующие осложнения:
- Стридозное дыхание из-за обструкции дыхательного пути. Ларингомаляция или даже асфиксия во сне.
- Психомоторное развитие ребенка сильно отстает от сверстников.
- Физическое развитие также отстает.
- Речь у больных нарушена.
- Частые инфекционные заболевания уха, которые становятся хроническими и приводят к нарушениям слуха.
- Синдром обструктивного апноэ, наступление смерти во сне варьирует в 14-91% случаев.
- Проблемы с зубами.
[25], [26], [27], [28], [29], [30]
Диагностика Синдрома Пьера Робена
Диагностика синдрома Пьера Робена трудностей не вызывает. Она основывается на клинических проявлениях. Чтобы исключить другие патологии, очень важно проконсультироваться у генетика.
У детей с врожденной аномалией Робена с самого рождения нарушено дыхание из-за постоянного западания языка. Младенец беспокойно себя ведет, кожные покровы его синюшные, при вдохах из грудной клетки вырывается хрип. В процессе кормления могут наступать удушья. Диагноз можно поставить также по необычному внешнему виду ребенка – «птичьему лицу». Часто у больных развиваются и другие пороки: миопия, катаракта, патология мочеполовой системы, патология сердца, аномалии развития позвоночника.
По этим клиническим проявлениям поставить правильный диагноз специалисту не составит труда.
[31], [32], [33], [34], [35], [36]
Лечение Синдрома Пьера Робена
Лечение проводят сразу же после рождения ребенка с синдромом Пьера Робена. Если заболевание носит легкий характер, то для улучшения состояния больного необходимо постоянно держать ребенка вертикально или лежа на животике. Голову младенца нужно наклонять к груди. В процессе кормления не рекомендовано держать ребенка в горизонтальном положении, чтобы пища не попадала в дыхательные пути.
Если недостаточное развитие нижней части челюсти выражено довольно сильно, применяется оперативное вмешательство для выведения западающего языка в нормальное физиологическое положение. При тяжелых случаях язык подтягивают и фиксируют на нижней губе. При очень тяжелых случаях необходимо проведения трахеостомии, глоссопексии, дистракционного остеогенеза нижней челюсти.
Также применяется и консервативное лечение.
Лекарства
Фенобарбитал. Снотворный и седативный препарат, отличается противосудорожным эффектом. В каждой таблетке находится 100 мл фенобарбитала. Дозировка является индивидуальной, так как зависит от степени тяжести болезни и состояния ребенка. Пациентам с печеночной недостаточностью, гиперкинезом, анемией, миастенией, порфирией, сахарным диабетом, депрессией, непереносимостью компонентов препарат запрещен. При приеме возможны следующие симптомы: головокружение, астения, галлюцинации, агранулоцитоз, тошнота, пониженное артериальное давление, аллергия.
Клоназепам. Препарат, который назначается для лечения эпилепсии. В лекарстве находится активное вещество клоназепам, которое является производным бензодиазепина. Отличается противосудорожным, анксиолитическим и миорелаксирующим эффектом. Доза устанавливается лечащим врачом, но не должна превышать максимальной – 250 мкг в день. Не принимать при бессоннице, мышечном гипертонусе, психомоторном возбуждении, панических расстройствах. При приеме возможны следующие симптомы: заторможенность, тошнота, дисменорея, головная боль, лейкопения, задержка или недержание мочи, алопеция, аллергия.
Сибазон. Выпускается в форме раствора и ректальных таблеток. Активным веществом является производным бензодиазепина (сибазон). Отличается седативным, анксиолитическим, противосудорожным эффектом. Дозировка является индивидуальной. Пациентам с хронической гиперкапнией, миастенией, непереносимостью бензодиазепинов принимать препарат запрещено. При употреблении средства возможно развитие таких симптомов: тошнота, запоры, головная боль, головокружение, икота, недержание мочеиспускания, аллергия.
Кортексин лиофилизат. Препарат с ноотропным действием. В лекарстве находится комплекс полипептидных фракций растворимых в воде и глицин. Дозировка является индивидуальной и назначается лечащим врачом в соответствии с состоянием больного. Пациентам с непереносимостью кортексина принимать препарат запрещено. Средство может вызывать аллергические реакции.
Физиотерапевтическое лечение
Как правило, при легких стадиях синдрома проводится позиционная терапия, когда ребенка укладывают на живот в вертикальном положении до тех пор, пока сила тяжести не заставит нижнюю часть челюсти расти правильно.
Оперативное лечение
Оперативное лечение используется, в первую очередь, для коррекции глоссоптоза. Существует несколько методов:
- Поддерживание с помощью серебряной нити языка. Нить проводится через нижнюю часть десны и нижнюю губу. Метод носит название Дугласа.
- Метод Духамеля – толстую серебряную нить проводят через основание языка пациента и две щеки. Использовать не дольше тридцати дней.
- Ортопедические аппараты для вытягивания и фиксации языка.
- В возрасте одного года можно проводить операцию по устранению расщелины на небе.
Профилактика
Единственным методом профилактики синдрома Пьера Робена является устранение возможных негативных факторов в дородовом периоде развития плода, пренатальная диагностика.
[37], [38], [39], [40], [41], [42], [43]
Прогноз
Прогноз и течение заболевания – тяжелые. Чаще всего в первые же дни жизни при средней и тяжелой стадии заболевания наступает смерть (причина – асфиксия). Также риск летального исхода в первый год достаточно высокий из-за многочисленных инфекций.
У пациентов в возрасте после двух лет – прогноз благоприятный.
[44], [45]
Источник