Аномалия арнольд киари код мкб

Содержание
- Синонимы диагноза
- Описание
- Дополнительные факты
- Симптомы
- Причины
- Классификация
- Диагностика
- Лечение
- Прогноз
- Основные медицинские услуги
- Клиники для лечения
Другие названия и синонимы
Синдром Арнольда-Киари.
Названия
Название: Аномалия Киари.
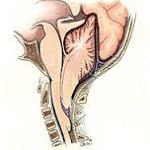
Аномалия Киари
Синонимы диагноза
Синдром Арнольда-Киари.
Описание
Аномалия Киари (мальформация Арнольда-Киари) — заболевание, при котором структуры головного мозга, расположенные в задней черепной ямке, опущены в каудальном направлении и выходят через большое затылочное отверстие. В зависимости от типа аномалия Киари может проявляться головной болью в затылке, болью в шейном отделе, головокружением, нистагмом, обмороками, дизартрией, мозжечковой атаксией, парезом гортани, снижением слуха и ушным шумом, нарушением зрения, дисфагией, дыхательными апноэ, стридором, расстройствами чувствительности, гипотрофией мышц и тетрапарезом. Аномалия Киари диагностируется путем проведения МРТ головного мозга, шейного и грудного отделов позвоночника. Аномалия Киари, сопровождающаяся стойким болевым синдромом или неврологическим дефицитом, подлежит хирургическому лечению (декомпрессия задней черепной ямки или шунтирующие операции).
Аномалия Киари
Дополнительные факты
В области соединения черепа с позвоночным столбом находится большое затылочное отверстие, на уровне которого ствол головного мозга переходит в спинной мозг. Выше этого отверстия локализуется задняя черепная ямка. В ней расположен мост, продолговатый мозг и мозжечок. Аномалия Киари связана с выходом части анатомических структур задней черепной ямки в просвет большого затылочного отверстия. При этом происходит сдавление находящихся в этой области структур продолговатого и спинного мозга, а также нарушение оттока цереброспинальной жидкости из головного мозга, приводящее к гидроцефалии. Вместе с платибазией, ассимиляцией атланта и тд аномалия Киари относится к врожденным порокам развития краниовертебрального перехода.
Аномалия Киари встречается по различным данным у 3-8 человек на 100 тысяч населения. В зависимости от типа аномалия Киари может диагностироваться в первые дни после рождения ребенка или стать неожиданной находкой у взрослого пациента. В 80% случаев аномалия Киари сочетается с сирингомиелией.
Симптомы
Боль в шее. Боль в шейном отделе позвоночника. Кашель. Рвота. Слабость мышц (парез).
Причины
До сих пор аномалия Киари остается заболеванием, об этиологии которого в неврологии нет единого мнения. Ряд авторов считает, что аномалия Киари связана с уменьшенным размером задней черепной ямки, приводящим к тому, что по мере роста расположенных в ней структур они начинают выходить через затылочное отверстие. Другие исследователи предполагают, что аномалия Киари развивается в результате увеличенных размеров головного мозга, который при этом как бы выталкивает содержимое задней черепной ямки через затылочное отверстие.
Спровоцировать переход незначительно выраженной аномалии в выраженную клиническую форму может гидроцефалия, при которой за счет увеличения желудочков увеличивается общий объем мозга. Поскольку аномалия Киари наряду с дисплазией костных структур краниовертебрального перехода сопровождается недоразвитием связочного аппарата этой области, любая черепно-мозговая травма может приводить к усугублению вклинения миндалин мозжечка в затылочное отверстие с манифестацией клинической картины заболевания.
Классификация
Аномалия Киари подразделяется на 4 типа:
Аномалия Киари I характеризуется опущением миндалин мозжечка ниже большого затылочного отверстия. Обычно она проявляется у подростков или во взрослом возрасте. Зачастую сопровождается гидромиелией — скоплением цереброспинальной жидкости в центральном канале спинного мозга.
Аномалия Киари II проявляется в первые дни после рождения. Кроме миндалин мозжечка при этой патологии через большое затылочное отверстие выходят также червь мозжечка, продолговатый мозг и IV желудочек. Аномалия Киари II типа намного чаще сочетается с гидромиелией, чем первый тип, и в подавляющем большинстве случаев связана с миеломенингоцеле — врожденной спинномозговой грыжей.
Аномалия Киари III отличается тем, что опустившиеся через большое затылочное отверстие мозжечок и продолговатый мозг, располагаются в менингоцеле шейно-затылочной области.
Аномалия Киари IV заключается в гипоплазии (недоразвитии) мозжечка и не сопровождается его смещением в каудальном направлении. Некоторые авторы относят эту аномалию к синдрому Денди-Уокера, при котором гипоплазия мозжечка сочетается с наличием врожденных кист задней черепной ямки и гидроцефалией.
Аномалия Киари II и Киари III часто наблюдается в комбинации с другими дисплазиями нервной системы: гетеротопией коры головного мозга, полимикрогирией, аномалиями мозолистого тела, кистами отверстия Можанди, перегибом сильвиевого водопровода, гипоплазией подкорковых структур, намета и серпа мозжечка.
Диагностика
Неврологический осмотр и стандартный перечень неврологических обследований (ЭЭГ, Эхо-ЭГ, РЭГ) не дают специфических данных, позволяющих установить диагноз «аномалия Киари». Как правило, они выявляют лишь признаки значительного повышения внутричерепного давления, т. Е. Гидроцефалию. Рентгенография черепа выявляет только костные аномалии, которыми может сопровождаться аномалия Киари. Поэтому до внедрения в неврологическую практику томографических методов исследования диагностика этого заболевания представляла для невролога большие затруднения. Теперь врачи имеют возможность поставить таким пациентам точный диагноз.
Следует отметить, что МСКТ и КТ головного мозга при хорошей визуализации костных структур краниовертебрального перехода не позволяют достаточно точно судить о мягкотканных образованиях задней черепной ямки. Поэтому единственным достоверным методом диагностики аномалии Киари на сегодняшний день является магнитно-резонансная томография. Ее проведение требует обездвиженности пациента, поэтому у маленьких детей она проводится в состоянии медикаментозного сна. Кроме МРТ головного мозга для выявления менингоцеле и сирингомиелических кист необходимо также проведение МРТ позвоночника, особенно его шейного и грудного отделов. При этом проведение МРТ исследований должно быть направлено не только на диагностику аномалии Киари, но и на поиск других аномалий развития нервной системы, которые часто с ней сочетаются.
Лечение
Бессимптомно протекающая аномалия Киари не нуждается в лечении. В случаях, когда аномалия Киари проявляется лишь наличием болей в шее и затылочной области, проводят консервативную терапию, включающую анальгетические, противовоспалительные и миорелаксирующие препараты. Если аномалия Киари сопровождается неврологическими нарушениями (парезы, расстройства чувствительности и мышечного тонуса, нарушения со стороны черепно-мозговых нервов и пр. ) или не поддающимся консервативной терапии болевым синдромом, то показано ее хирургическое лечение.
Наиболее часто в лечении аномалии Киари применяется краниовертебральная декомпрессия. Операция включает расширение затылочного отверстия за счет удаления части затылочной кости; ликвидацию сдавления ствола и спинного мозга за счет резекции миндалин мозжечка и задних половин двух первых шейных позвонков; нормализацию циркуляции цереброспинальной жидкости путем подшивания в твердую мозговую оболочку заплаты из искусственных материалов или аллотрансплантата. В некоторых случаях аномалия Киари лечится при помощи шунтирующих операций, направленных на дренирование цереброспинальной жидкости из расширенного центрального канала спинного мозга. Цереброспинальная жидкость может отводиться в грудную или брюшную полость (люмбоперитонеальное дренирование).
Прогноз
Важное прогностическое значение имеет тип, к которому относится аномалия Киари. В некоторых случаях аномалия Киари I может на протяжении всей жизни пациента сохранять бессимптомное течение. Аномалия Киари III в большинстве случаев приводит к летальному исходу. При появлении неврологических симптомов аномалии Киари I, а также при аномалии Киари II большое значение имеет своевременное проведение хирургического лечения, поскольку возникший неврологический дефицит плохо восстанавливается даже после успешно проведенной операции. По различным данным эффективность хирургической краниовертебральной декомпрессии составляет 50-85%.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
Содержание
- Описание
- Дополнительные факты
- Причины
- Классификация
- Диагностика
- Лечение
- Прогноз
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Q07,0 Синдром Арнольда-Киари.
Q07.0 Синдром Арнольда-Киари
Описание
Аномалия Киари (мальформация Арнольда-Киари) — заболевание, при котором структуры головного мозга, расположенные в задней черепной ямке, опущены в каудальном направлении и выходят через большое затылочное отверстие. В зависимости от типа аномалия Киари может проявляться головной болью в затылке, болью в шейном отделе, головокружением, нистагмом, обмороками, дизартрией, мозжечковой атаксией, парезом гортани, снижением слуха и ушным шумом, нарушением зрения, дисфагией, дыхательными апноэ, стридором, расстройствами чувствительности, гипотрофией мышц и тетрапарезом. Аномалия Киари диагностируется путем проведения МРТ головного мозга, шейного и грудного отделов позвоночника. Аномалия Киари, сопровождающаяся стойким болевым синдромом или неврологическим дефицитом, подлежит хирургическому лечению (декомпрессия задней черепной ямки или шунтирующие операции).
Дополнительные факты
В области соединения черепа с позвоночным столбом находится большое затылочное отверстие, на уровне которого ствол головного мозга переходит в спинной мозг. Выше этого отверстия локализуется задняя черепная ямка. В ней расположен мост, продолговатый мозг и мозжечок. Аномалия Киари связана с выходом части анатомических структур задней черепной ямки в просвет большого затылочного отверстия. При этом происходит сдавление находящихся в этой области структур продолговатого и спинного мозга, а также нарушение оттока цереброспинальной жидкости из головного мозга, приводящее к гидроцефалии. Вместе с платибазией, ассимиляцией атланта и тд аномалия Киари относится к врожденным порокам развития краниовертебрального перехода.
Аномалия Киари встречается по различным данным у 3-8 человек на 100 тысяч населения. В зависимости от типа аномалия Киари может диагностироваться в первые дни после рождения ребенка или стать неожиданной находкой у взрослого пациента. В 80% случаев аномалия Киари сочетается с сирингомиелией.
Q07.0 Синдром Арнольда-Киари
Причины
До сих пор аномалия Киари остается заболеванием, об этиологии которого в неврологии нет единого мнения. Ряд авторов считает, что аномалия Киари связана с уменьшенным размером задней черепной ямки, приводящим к тому, что по мере роста расположенных в ней структур они начинают выходить через затылочное отверстие. Другие исследователи предполагают, что аномалия Киари развивается в результате увеличенных размеров головного мозга, который при этом как бы выталкивает содержимое задней черепной ямки через затылочное отверстие.
Спровоцировать переход незначительно выраженной аномалии в выраженную клиническую форму может гидроцефалия, при которой за счет увеличения желудочков увеличивается общий объем мозга. Поскольку аномалия Киари наряду с дисплазией костных структур краниовертебрального перехода сопровождается недоразвитием связочного аппарата этой области, любая черепно-мозговая травма может приводить к усугублению вклинения миндалин мозжечка в затылочное отверстие с манифестацией клинической картины заболевания.
Классификация
Аномалия Киари подразделяется на 4 типа:
Аномалия Киари I характеризуется опущением миндалин мозжечка ниже большого затылочного отверстия. Обычно она проявляется у подростков или во взрослом возрасте. Зачастую сопровождается гидромиелией — скоплением цереброспинальной жидкости в центральном канале спинного мозга.
Аномалия Киари II проявляется в первые дни после рождения. Кроме миндалин мозжечка при этой патологии через большое затылочное отверстие выходят также червь мозжечка, продолговатый мозг и IV желудочек. Аномалия Киари II типа намного чаще сочетается с гидромиелией, чем первый тип, и в подавляющем большинстве случаев связана с миеломенингоцеле — врожденной спинномозговой грыжей.
Аномалия Киари III отличается тем, что опустившиеся через большое затылочное отверстие мозжечок и продолговатый мозг, располагаются в менингоцеле шейно-затылочной области.
Аномалия Киари IV заключается в гипоплазии (недоразвитии) мозжечка и не сопровождается его смещением в каудальном направлении. Некоторые авторы относят эту аномалию к синдрому Денди-Уокера, при котором гипоплазия мозжечка сочетается с наличием врожденных кист задней черепной ямки и гидроцефалией.
Аномалия Киари II и Киари III часто наблюдается в комбинации с другими дисплазиями нервной системы: гетеротопией коры головного мозга, полимикрогирией, аномалиями мозолистого тела, кистами отверстия Можанди, перегибом сильвиевого водопровода, гипоплазией подкорковых структур, намета и серпа мозжечка.
Диагностика
Неврологический осмотр и стандартный перечень неврологических обследований (ЭЭГ, Эхо-ЭГ, РЭГ) не дают специфических данных, позволяющих установить диагноз «аномалия Киари». Как правило, они выявляют лишь признаки значительного повышения внутричерепного давления, т. Е. Гидроцефалию. Рентгенография черепа выявляет только костные аномалии, которыми может сопровождаться аномалия Киари. Поэтому до внедрения в неврологическую практику томографических методов исследования диагностика этого заболевания представляла для невролога большие затруднения. Теперь врачи имеют возможность поставить таким пациентам точный диагноз.
Следует отметить, что МСКТ и КТ головного мозга при хорошей визуализации костных структур краниовертебрального перехода не позволяют достаточно точно судить о мягкотканных образованиях задней черепной ямки. Поэтому единственным достоверным методом диагностики аномалии Киари на сегодняшний день является магнитно-резонансная томография. Ее проведение требует обездвиженности пациента, поэтому у маленьких детей она проводится в состоянии медикаментозного сна. Кроме МРТ головного мозга для выявления менингоцеле и сирингомиелических кист необходимо также проведение МРТ позвоночника, особенно его шейного и грудного отделов. При этом проведение МРТ исследований должно быть направлено не только на диагностику аномалии Киари, но и на поиск других аномалий развития нервной системы, которые часто с ней сочетаются.
Лечение
Бессимптомно протекающая аномалия Киари не нуждается в лечении. В случаях, когда аномалия Киари проявляется лишь наличием болей в шее и затылочной области, проводят консервативную терапию, включающую анальгетические, противовоспалительные и миорелаксирующие препараты. Если аномалия Киари сопровождается неврологическими нарушениями (парезы, расстройства чувствительности и мышечного тонуса, нарушения со стороны черепно-мозговых нервов и пр. ) или не поддающимся консервативной терапии болевым синдромом, то показано ее хирургическое лечение.
Наиболее часто в лечении аномалии Киари применяется краниовертебральная декомпрессия. Операция включает расширение затылочного отверстия за счет удаления части затылочной кости; ликвидацию сдавления ствола и спинного мозга за счет резекции миндалин мозжечка и задних половин двух первых шейных позвонков; нормализацию циркуляции цереброспинальной жидкости путем подшивания в твердую мозговую оболочку заплаты из искусственных материалов или аллотрансплантата. В некоторых случаях аномалия Киари лечится при помощи шунтирующих операций, направленных на дренирование цереброспинальной жидкости из расширенного центрального канала спинного мозга. Цереброспинальная жидкость может отводиться в грудную или брюшную полость (люмбоперитонеальное дренирование).
Прогноз
Важное прогностическое значение имеет тип, к которому относится аномалия Киари. В некоторых случаях аномалия Киари I может на протяжении всей жизни пациента сохранять бессимптомное течение. Аномалия Киари III в большинстве случаев приводит к летальному исходу. При появлении неврологических симптомов аномалии Киари I, а также при аномалии Киари II большое значение имеет своевременное проведение хирургического лечения, поскольку возникший неврологический дефицит плохо восстанавливается даже после успешно проведенной операции. По различным данным эффективность хирургической краниовертебральной декомпрессии составляет 50-85%.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
Общие сведения
Синдром Арнольда-Киари является пороком развития мозжечка – отдела головного мозга, отвечающего за координацию, мышечный тонус и равновесие. Патологии присвоен код по МКБ-10 Q07.0 и она представляет собой опущение миндалин мозжечка вниз на уровень первого, а порой второго шейного позвонка (ниже черты Чемберлена) и блокирует нормальный ток спинномозговой жидкости.
Заболевание чаще всего сочетается с микрогирией, сдавливанием заднего отдела мозга, стенозом водопровода мозга, базилярной импрессией, инвагинацией, недоразвитием четверохолмия и другими мальформациями нервной системы. Синдром чаще всего встречается у особ в возрасте 12-71 год и не превышает 000,9%.

Локализация и строение мозжечка
Патогенез
В основе патофизиологии обычно лежит несоответствие размеров задней черепной ямки и имеющихся в ней структур нервной системы, а также:
- развитие аномалий тел шейных позвонков, включая их расщепление чаще всего первого (данный механизм развития встречается в 5% случаев), ассимиляцию атланта — сращивание шейного позвонка с затылочной костью;
- смещение структур мозжечка в период бурного роста мозга при медленно растущих костях черепа;
- гидроцефалия – избыточное скопление цереброспинальной жидкости;
- сирингомиелия – анормальный процесс развития полостей в спинном мозге;
- миеломенингоцеле – врожденный дефект развития нервной трубки;
- различные врожденные заболевания, в том числе платибазия, аномалия Денди-Уокера.
Классификация
В зависимости от клинической картины и степени развития анатомических аномалий синдром Арнольда-Киари бывает четырех типов.
Аномалия Арнольда Киари 1 типа – это классический вариант порока развития мозжечка
Синдром Арнольда-Киари 1 типа проявляется в виде проникновения миндалин мозжечка в полость позвоночного канала, вызывающего гидромиелию и опущение структур задней черепной ямки ниже большого затылочного отверстия на 3-5 мм и более, причем нет никаких других мальформаций нервной системы. Средняя продолжительность жизни обычно не превышает 25-40 лет.
Порок мозжечка 2 типа
Представляет собой опущение в полость позвоночного канала различных структур мозжечка и тканей ствола, при этом данная нейропозвоночная мальформация сочетается с миеломенингоцеле (врожденной спинномозговой грыжей) и гидроцефалией. Манифестация происходит практические сразу после рождения.
Мальформация Арнольда Киари 3 типа
Мальформация отличается наличием затылочного энцефалоцеле и различных признаков аномалии второго типа. Обычно не совместима с жизнью.
Четвертый тип Синдром Арнольда — Киари
По своей сути это аплазия либо гипоплазия всех или отдельных структур мозжечка, то есть бывает тотальной и субтотальной. Первый вариант встречается достаточно редко и сочетается с прочими тяжёлыми аномалиями и заболеваниями нервной системы, включая анэнцефалию, амиелию. При субтотальной агенезии наблюдаются пороки развития других участков головного мозга, например агенезия моста, отсутствие четвёртого желудочка и пр.
Гипоплазия мозжечка встречается в форме уменьшения всего мозжечка или охватывает отдельные части, при этом сохраняются нормальные структуры без утраты функций. Встречается одно- и двусторонняя, лобарная, лобулярная и интракортикальная гипоплазия. Изменения конфигурации листков мозжечка обычно представлено в виде аллогирии, полигирии или агирии.
Кроме того, некоторые авторы выделяют два дополнительных типа:
- тип «0» — заболевание имеет сходную клиническую картину с синдромом Арнольда Киари, но без анатомических изменений опущения миндалин или гипоплазии мозжечка;
- тип «1,5» — у больных выявляется опущение не только миндалин мозжечка в затылочное отверстие, но и ствола головного мозга.
Причины
Помимо роли наследственного и генетического фактора существует несколько теорий возникновения пороков мозжечка. Традиционная теория говорит, что опущение миндалин вызвано натяжением струны спинного мозга в результате напряжения концевой нити при развитии той или иной мальформации. Исключением становится болезнь Киари 1 типа, ведь единственным нарушением в этом случае становится опущение миндалин и оно может быть спровоцировано:
- гидродинамическими явлениями — нарушением циркуляции спинно-мозговых жидкостей;
- черепно-мозговыми и родовыми травмами;
- мальформацией — маленькие размеры и ограниченность затылочного отверстия могут приводить к опущению миндалин в просвет позвоночного канала;
- анормально натянутой связкой — так называемой концевой нитью (по теории доктора М.Б. Ройо Сальвадора — Filum System).
Кроме того, ученые выделяют ряд факторов, которые могут повысить риск развития аномалии Арнольда-Киари первого типа:
- генетическая предрасположенность – наличие патологии у родителей и более дальних предков, хотя хромосомных аномалий до сих пор не выявлено;
- травмы, особенно падения, могут вызвать компрессию и усиление натяжения концевой нити и привести к опущению миндалин мозжечка;
- неправильное поведение и вредные привычки женщины в период вынашивания младенца — злоупотребление и хаотичный прием медикаментов, курение, употребление алкоголя, а также перенесенные вирусные заболевания.
Симптомы
Симптоматика при различных типах синдрома Арнольда Киари может существенно отличаться – все зависит от степени натяжения и компрессии нервных структур затылочного отдела, но чаще всего у больных наблюдаются:
- головные боли;
- прогрессирующее увеличение размеров окружности головы;
- периодические боли в различных отделах позвоночника;
- парез и боль в различных областях конечностей;
- нарушения зрения и чувствительности, включая дизестезии и парестезии;
- шумы в ушах;
- паралич лицевого, глазодвигательного нерва;
- дрожь;
- приступы головокружения;
- бессонница;
- рвота;
- обмороки;
- нистагм;
- апноэ;
- нарушение работы сфинктеров, мышц языка, глотки, онемение и различные проявления мышечной слабости;
- нарушения способности глотания (дисфагия);
- нарушения памяти;
- несогласованность движений (атаксия) и как следствие — неуклюжая походка, причем нарушения наиболее ярко выражены на этапе формирования;
- сколиоз;
- трудности с удержанием позиций тела и равновесия, а также координации;
- затруднения при желании выразить мысль и подобрать слова.
Проявления синдрома Арнольда-Киари хронические, имеют тенденцию увеличивать интенсивность, что с каждым разом существенно ухудшает состояние больного и ограничивает его привычный образ жизни.
Самое опасное, что аномалия Арнольда-Киари может привести к внезапной смерти, ведь спинно-мозговые центры отвечают за сердечно-дыхательные функции, а давление миндалин мозжечка на них может спровоцировать остановку дыхания — апноэ, которое станет причиной летального исхода.
Симптомы, которые вызывает аномалия Арнольда Киари 1 степени
Патология 1 степени обычно выявляется случайно во время проведения МРТ, ведь больные помимо эпизодов апноэ и обмороков могут испытывать только:
- боль в шейно-затылочном отделе;
- снижение чувствительности.
Анализы и диагностика
При первых признаках необходимо пройти неврологический осмотр и провести оценку выраженности клинико-функциональных нарушений. Для подтверждения диагноза чаще всего применяются:
- нейровизуализационной методики, наиболее предпочтительно МРТ;
- электроэнцефалография;
- рентгенография черепа;
- миелография, которая позволяет выявить дефекты верхнего шейного отдела спинного мозга, ствола мозга, а также локализацию мозжечка ниже черты Чемберлена в области затылочного отверстия.

Результаты МРТ при аномалии Арнольда Киари с сирингомиелической кистой и в норме
Лечение
Тактика лечения при различных типах пороков развития мозжечка Арнольда-Киари обычно является консервативной либо продумывается нейрохирургом и представляет собой декомпрессию, наложение шунта (при выраженной гидроцефалии) или краниотомию затылочного отверстия.
Однако, благодаря докторской диссертации доктора М.Б. Ройо Сальвадора была разработана новаторская техника этиологического лечения — Filum System, которая направлена на устранение причины заболевания и патологического механизма натяжения путем хирургического минимально инвазивного рассечения концевой нити. Преимуществом методики является возможность остановить болезнь Арнольда Киари при минимальных рисках (смертность – 0%), главное выявить мальформацию и провести операцию как можно раньше. Она обычно занимает не более 45 минут и позволяет добиться симптоматического улучшения состояния, а в отдельных случаях даже поднятия миндалин мозжечка. Несмотря на короткий восстановительный период, методика имеет ряд недостатков:
- после операции остается небольшой шов;
- может возникать субъективное ощущение снижения силы конечностей;
- улучшение мозгового кровообращения вначале постоперативного периода может вызвать перепады настроения.
Но это незначительные неудобства по сравнению с минусами затылочной краниотомии, у которой:
- смертность 1-12%;
- причина заболевания не устраняется, поэтому улучшения сохраняются непродолжительный период;
- последствия оперативного вмешательства могут быть очень серьёзными и непредсказуемыми, включая отек мозга, дальнейшее опущение миндалин мозжечка, усугубление неврологической симптоматики, гемодинамические нарушения, гидроцефалию, пневмоэнцефалию, внутримозговые кровоизлияния, тетрапарез, неврологический дефицит и т.д.
Доктора
Лекарства
Медикаментозная терапия обычно симптоматическая, чаще всего пациентам назначают НПВС, миорелаксанты и анальгетики. Например, Прегабалин, Карбамазепин, Спазмалгон, Палексия и пр.
Проблемой становится неэффективность противовоспалительных и обезболивающих препаратов при купировании приступов головной боли или других симптомах.
Процедуры и операции
- лечебная гимнастика (кинезотерапия);
- избирательный точечный массаж;
- иглорефлексотерапия и электроакупунктура при наличии мышечной спастичности и болевых синдромов;
- занятия с логопедом (при необходимости коррекции нарушений речи);
- психокоррекционная работа психолога при наличии эмоциональных, дисфорических или депрессивных расстройств.
У детей
Чаще всего отмечается блокада внутри желудочковой системы и представляет собой несообщающуюся гидроцефалию. Это обычно вызвано сужением сильвиева водопровода либо недоразвитием отверстий Мажанди и Люшки. Как генерализованное нарушение развития нервной системы, оно может сочетаться с микро- или макрогирией, порэнцефалией, агенезией мозолистого тела, слиянием полушарий мозга, агенезией червя мозжечка, позвоночных расщелин, менингоцеле, энцефалоцеле, сирингомиелией, гидромиелией. Причиной сообщающейся гидроцефалии может быть процесс образования спаек в субарахноидальной области в основания мозга, перинатальные субарахноидальные или внутрижелудочковые кровоизлияния.
Синдром у детей может возникать в результате травматических повреждений клиновидно-затылочной и клиновидно-решетчатой области при родовых травмах. Клинические проявления сводятся к нарушению кормления (рефлюксу и аспирации), эпизодам апноэ, стридору, нарушениям краниальной иннервации, судорожным приступам устойчивым к противосудорожным препаратам.
Синдром Арнольда Киари у плода
Патология Киари является врожденным остеоневропатией и формируется на этапе закладки и развития нервной трубки у плода, асинхронным формированием нервного ствола и костей черепной ямки.
Морфологические проявления данной патологии можно выявить во время проведения ультразвукового исследования, предопределить дальнейший прогноз. Чаще всего выявление патологии становится причиной для прерывания беременности.
Список источников
- Петровский Б.В. Большая медицинская энциклопедия — М.:: Советская энциклопедия, 1981. — Т. XV (Меланома-Мудров). — С. 576.
- Гусев Е. И., Коновалов А. Н., Скворцова В. И. Неврология и нейрохирургия. — М.: ГЭОТАР-Медиа, 2010.— 624 с.
Источник