Анализ синдром ломкой х хромосомы

[42-073]
Генодиагностика синдрома ломкой Х-хромосомы (синдром Мартина — Белл)
3740 руб.
Увеличение числа CGG-триплетных повторов (экспансия) в 5-нетранслируемой части гена FMR1 является причиной развития синдрома ломкой Х-хромосомы (синдром Мартина — Белл), Х-сцепленного наследственного заболевания.
Синонимы русские
Ломкая Х-хромосома (с. Мартина — Белл), ген FMR1, экспансия триплетных повторов, генетическое обследование.
Синонимы английские
FMR1-Related Disorders, fragile X syndrome, gene FMR1, expansion of CGG (cytosine- guanine -guanine) triplet repeats.
Название гена
Ген FMR1.
Локализация гена на хромосоме
Локус Xq27.3.
Метод исследования
Полимеразная цепная реакция (ПЦР) — Фрагментный анализ гена FMR1.
Какой биоматериал можно использовать для исследования?
Венозную кровь.
Как правильно подготовиться к исследованию?
- Не курить в течение 30 минут до исследования.
Общая информация об исследовании
Синдром ломкой Х-хромосомы (синдром Мартина — Белл — СМБ) представляет собой наиболее частую моногенную причину нарушения развития у детей, а также наиболее частую известную моногенную причину развития аутизма. Данное наследственное заболевание характеризуется увеличением количества CGG-повторов в 5-нетранслируемой части FMR1-гена, располагающегося на Х-хромосоме и кодирующего одноименный белок синдрома ломкой Х-хромосомы (БХЛХ). Данная генетическая аберрация приводит к гипермителированию зоны экспансии, а также прилежащего промоутера FMR1-гена. Гипермителирование ингибирует транскрипцию гена и нарушает синтез белка БХЛХ.
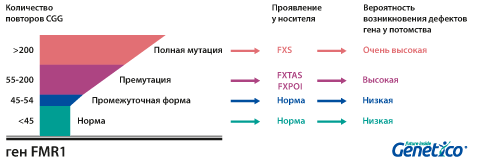
В норме количество повторов не более 59. У пациентов с СМБ количество CGG-триплетов обычно превышает 200. Распространенность данного заболевания у мужчин составляет 1:3600, у женщин – 1:4000.
Заболевание СМБ является Х-сцепленным и наследуется по доминантному типу, то есть имеется 50% риска его наследования от больной матери. Для СМБ нехарактерен феномен антиципации. Размер экспансии не коррелирует с тяжестью симптоматики, уровнем пенетрантности и временем первых проявлений заболевания.
БХЛХ является РНК-связывающимся протеином, способным образовывать рибонуклеопротеиновые комплексы, связываться с полисомами и ингибировать трансляцию многих белков. Считается, что белок БХЛХ участвует в эмбриональном развитии органов ЦНС, яичников, а также в нормальном синтезе белков локально в дендритах нейронов. Экспансия CGG-повторов ингибирует транскрипцию FMR1-гена и нарушает синтез белка БХЛХ.
Клинические проявления:
СМБ может проявляться в виде несиндромального отставания в развитии.
- Отставание в развитии, гиперактивность, синдром дефицита внимания.
- Задержка психоречевого развития с элементами аутизма.
- Черепно-лицевой дисморфизм — вытянутое лицо, выступающий подбородок, большие оттопыренные уши, страбизмус, широкий лоб, высокое небо.
- Гиперподвижность суставов, пролапс митрального клапана.
- Макроорхидизм.
- Гипотония мышц.
- Pectus excavatum — деформация грудной клетки.
- Судорожный синдром.
Для чего используется исследование?
В соответствии с международными клиническими рекомендациями, генетическое обследование на синдром ломкой Х-хромосомы проводится при наличии у пациента клинической симптоматики, характерной для данного заболевания, а также родственникам и детям больного.
Когда назначается исследование?
- Дифференциальный диагноз отставания развития;
- при подозрении на синдром ломкой Х-хромосомы;
- при когнитивных и нейропсихических нарушениях;
- при раннем выявлении заболевания у родственников;
- при планировании семьи.
Что означают результаты?
Генетическое обследование является основным методом подтверждения диагноза и основано на подсчете числа тройных CGG-повторов с помощью метода фрагментного анализа в гене FMR1. Диагностическая значимость обнаруженного числа CGG-повторов в гене FMR1 представлена в таблице:
Количество CGG-повторов | Диагноз и прогноз |
5-44 — нормальные аллели | Диагноз «СМБ» исключен (> 99%). Риск развития СМБ у следующих поколений крайне низок |
45-59 — умеренное увеличение | Диагноз «СМБ» исключен (> 99%). Имеется вероятность развития СМБ через поколение или у дальних родственников |
60-199 — предэкспансия | Возможен легкий фенотип СМБ. Повышенный риск развития ПЯН и СТА, ассоциированных с СЛХХ. Имеется вероятность развития СМБ у следующего поколения (зависит от размера экспансии — 3% для материнской аллели размером 55-59 и ~ 100% для аллели 90 CGG и более) |
≥ 200 — выраженная экспансия | Диагноз «СМБ» подтвержден. 50% вероятности передачи СМБ следующему поколению матерью |
Что может влиять на результат?
Хотя генетический тест является точным методом лабораторной диагностики, время клинических проявлений заболевания (пенетрантность болезни) зависит от внешней среды, индивидуальных генетических факторов. Для оценки характера наследования у детей и родственников, характера развития заболевания в последующем, назначения лечения рекомендуется получить консультацию специалиста.
Важные замечания
- Для получения заключения по результату обследования необходимо проконсультироваться у клинического генетика.
Кто назначает исследование?
Невролог, психиатр, врач-генетик.
Также рекомендуется
[16-001] Исследование кариотипа (количественные и структурные аномалии хромосом) по лимфоцитам периферической крови (1 человек)
Литература
- Saul RA, Tarleton JC. FMR1-Related Disorders. 1998 Jun 16 [Updated 2012 Apr 26]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018.
- D’Hulst C, Kooy RF Fragile X syndrome: from molecular genetics to therapy Journal of Medical Genetics 2009;46:577-584.
- Pugin A, Faundes V, Santa María L, Curotto B, Aliaga S, Salas I, et al. Clinical, molecular, and pharmacological aspects of FMR1-related disorders. Neurol (English Ed 2017;32:241–52.
- Wheeler AC, Bailey Jr DB, Berry-Kravis E, Greenberg J, Losh M, Mailick M, et al. Associated features in females with an FMR1 premutation. J Neurodev Disord 2014;6:30.
Источник
Генетические особенности синдрома ломкой Х-хромосомы
Среди группы наследственных болезней есть два заболевания, относящихся к самым частым причинам интеллектуальной недостаточности. Самая известная и наиболее распространённая патология – синдром Дауна, связанный с наличием лишней 21-ой хромосомы в геноме человека. В этой статье мы расскажем о втором по распространенности наследственном заболевании, которое приводит к умственной отсталости, а также может сопровождаться другими клиническими проявлениями.
Синдром ломкой X-хромосомы или синдром Мартина-Белл является результатом нарушения в гене FMR1 (fragile X mental retardation-1), который расположен на Х-хромосоме и играет важную роль в появлении и развитии нервных связей, обучении и запоминании. Частота этого синдрома среди мальчиков составляет 1:4000.
Так называемая «ломкость» X-хромосомы проявляется в том, что хромосома выглядит нетипично при специальном окрашивании, как будто один кусок отделился, хотя физически она остается цельной. Генетическая основа этого явления заключается в увеличении числа тринуклеотидных повторов CGG в гене FMR1, расположенном на X-хромосоме.
У здоровых людей число повторов в этом гене колеблется от 5 до 54. Если повторов больше 200, то наработка белка с гена FMR1 нарушается, что приводит к развитию синдрома Мартина-Белл и клиническому проявлению заболевания. Премутационное состояние — это количество повторов CGG от 55 до 200. В таком состоянии заболевание у людей в типичной форме не проявляется, но чем больше повторов в этом гене у носителя, тем больше вероятность того, что у ее или его детей количество повторов будет больше 200 и заболевание разовьется. В случае носительства премутации при формировании половых клеток количество повторов может увеличиваться, поэтому если у родителя количество повторов от 55 до 200, то высока вероятность рождения ребенка с мутантным геном FMR1 и синдромом Мартина-Белл. При этом носительство премутационного состояния будущим папой и мамой неравнозначно по вероятности возникновения мутантного аллеля у их детей: если носитель – мама, то вероятность значительного увеличения числа повторов гораздо выше. Количество повторов от 45 до 54 является промежуточной формой, которая не имеет никакого влияния на здоровье человека, но может приводить к проблемам у будущих поколений, как и в случае премутационного состояния гена.
Важно учитывать, что наследование и развитие заболевания зависит от пола, так как ген FMR1 находится на Х-хромосоме. У мужчин только одна Х-хромосома, которую они получают от матери. Поэтому, в случае, если эта одна хромосома оказалась «ломкой», у них проявляется заболевание. У женщин две Х-хромосомы, однако активно работает только одна из них. Поэтому наличие одной Х-хромосомы с мутантным геном FMR1 может не проявляться клинически, в случае инактивации именно «ломкой» хромосомы, или приводить к развитию заболевания в 30-50% случаев. Мужчина с ломкой Х-хромосомой может передать её всем дочерям, но ни одному из сыновей. Женщина с мутантной хромосомой имеет шансы передать её как сыновьям, так и дочерям с равной вероятностью.
Премутационное состояние гена влияет как на судьбу потомков носителя такого гена, так и непосредственно на его здоровье:
Развитие первичной недостаточности яичников (FXPOI) (снижение овариального резерва и наступление менопаузы до 40 лет). Мутация FMR1 является причиной преждевременного истощения яичников у 5% женщин с этим диагнозом. Среди носительниц премутации примерно у четверти развивается это состояние. Оно влияет не только на общие репродуктивные возможности, но и на подбор протокола стимуляции при ВРТ, так как часто оказывается причиной бедного ответа яичников на стимуляцию. Интересно, что по данным, полученным в центре Genetico, хотя бедный ответ яичников на стимуляцию влияет на число получаемых в цикле эмбрионов, он не приводит к увеличению доли анеуплоидных эмбрионов.
Тремор/атаксия, ассоциированные с ломкой Х-хромосомой (FXTAS). Это состояние чаще развивается у мужчин: при носительстве премутации мужчиной проявляется в 33% случаев, а при носительстве премутации женщиной – лишь в 5-10%. Синдром FXTAS начинает проявляться в пожилом возрасте. Наблюдается тремор, шаткая походка, может страдать речь.
Метод диагностики, используемый в лаборатории Genetico, основан на использовании полимеразной цепной реакции с особым набором праймеров, позволяющих не только детектировать нормальное, премутационное и мутационное состояния, но и точно определить количество повторов в случаях, когда их меньше 200. Такая диагностика позволяет выявить синдром ломкой X-хромосомы на молекулярном уровне, а также оценить вероятность рождения ребенка с этим синдромом и возможность развития у пациента расстройств, связанных с увеличенным количеством повторов в гене FMR1. Такая диагностика также позволяет детектировать наличие AGG повторов среди повторов CGG. Полагают, что участки AGG, прерывающие длинную последовательность из CGG повторов, придают ДНК устойчивость и снижают риск увеличения количества повторов в следующем поколении.
Генетический тест, определяющий количество повторов в гене FMR1, рекомендуется пройти в первую очередь женщинам с синдромом преждевременного истощения яичников или с выявленной неслучайной инактивацией Х-хромосомы (косвенный признак), семьям, в которых есть сыновья с интеллектуальной недостаточностью. Также анализ состояния гена FMR1 необходим:
1) женщинам с репродуктивными проблемами или нарушениями фертильности, связанными с повышенным уровнем фолликулостимулирующего гормона (ФСГ)
2) пациентам с интеллектуальной недостаточностью и их родственникам
3) тем, у кого в семье были случаи синдрома ломкой Х-хромосомы или умственной отсталости без точного диагноза
4) женщинам, у родственников которых наблюдались нарушения, связанные с премутационным состоянием FMR1
5) пациентам с поздно проявившимся тремором и мозжечковой атаксией (нарушения согласованности работы мышц из-за поражения систем мозга, управляющих движением мышц).
В случае обнаружения бессимптомного носительства мутации в гене FMR1 у женщины может быть рекомендовано использование донорских ооцитов или проведение преимплантационной генетической диагностики (ПГД) с целью исключить возможность проявления синдрома у ребенка. Также важно правильно оценивать риск рождения больного ребенка в случае премутационного состояния гена FMR1 у будущих родителей. В таком случае по результатам теста рекомендуется консультация врача-генетика.
Автор: Очир Мигяев
Стажер лаборатории Genetico
Источник
Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 29 декабря 2019;
проверки требуют 3 правки.
Синдро́м Ма́ртина — Белл (синдром ломкой X-хромосомы, fragile X syndrome, FraX (от англ. fragile — хрупкий, ломкий)) — наследственное заболевание.
Развитие синдрома связано с экспансией единичных тринуклеотидов (ЦГГ) в Х-хромосоме и приводит к недостаточной экспрессии белка FMR1, который необходим для нормального развития нервной системы. Существует четыре основных состояния хромосомного участка, подверженного нарушениям при синдроме ломкой Х-хромосомы, которые относятся к удлинению повторяющихся последовательностей ЦГГ. Нормальное количество повторов (отсутствие синдрома) — от 29 до 31. Премутация — от 55 до 200 повторов (синдром не развивается). Полная мутация — более 200 повторов (обычно от 230 до 4000), при которой проявляется синдром. Промежуточное состояние, или аллели серой зоны, — от 40 до 60 повторов[2].
История[править | править код]
В начале ХХ века учёные заметили преобладание умственной отсталости у лиц мужского пола. В 1943 году Джеймсом Мартином (James Purdon Martin) и Джулией Белл впервые была описана семья, где умственная отсталость наследовалась сцепленно с полом[3]. В этой английской семье 11 умственно отсталых детей мужского пола с психическим возрастом 2—4 года родились у интеллектуально нормальных матерей[3]. В двух случаях относительно небольшой интеллектуальный дефицит был у женщин этого семейства[3]. В 1969 году Hebert Lubs, проводя цитогенетическое обследование, выявил у больного синдромом Мартина-Белл вторичную перетяжку на длинном плече Х-хромосомы в локусе Xq27-28.
Частота распространения — 1:1000—1:2000 новорожденных мальчиков.
Этиология[править | править код]
Положение гена «FMR1» на Х-хромосоме.
Синдром ломкой Х-хромосомы развивается в результате мутации гена FMR1 в Х-хромосоме. Мутация в этом гене встречается приблизительно у одного из 2000 мужчин и у одной из 259 женщин. Распространённость непосредственно заболевания — приблизительно 1 из 4000 мужчин и 6000 женщин[4].
Экспансия повторяющихся кодонов ЦГГ приводит к гиперметилированию ДНК в промоторе гена FMR1 и, как следствие, фактическому прекращению его экспрессии.
Как предполагают, аномальное метилирование промотора гена FMR1 в локусе Хq27.3 является причиной формирования сайта ломкости Х-хромосомы. По этому цитогенетическому признаку синдром Мартина — Белл получил своё второе название — синдром ломкой Х-хромосомы.
Мутация гена FMR1 приводит к подавлению транскрипции белка FMR1. У здоровых индивидов FMR1, как считают, регулирует значительную популяцию мРНК: FMR1 играет важную роль в обучении и запоминании, а также принимает участие в развитии аксонов, формировании синапсов, появлении и развитии нервных связей[5].
Наследование[править | править код]
Синдром ломкой Х-хромосомы — сцепленное с полом доминантное заболевание с редуцированной пенетрантностью[6].
Мужчины имеют одну Х-хромосому, соответственно, если она содержит мутантный аллель, у носителя развивается заболевание. Женщины несут две Х-хромосомы, таким образом, их шанс получить нормальный аллель удваивается. Женщина с мутантным геном FMR1 может иметь симптомы болезни или быть здоровой. Несмотря на то, что вторая Х-хромосома может служить резервной копией, только одна Х-хромосома активна в каждой отдельной клетке, вследствие инактивации второй.
Мужчина с ломкой Х-хромосомой не может передать её ни одному из сыновей, только всем дочерям. Женщина с одной мутантной хромосомой имеет одинаковые шансы передать её как дочерям, так и сыновьям с вероятностью 50 %. Наследование синдрома ломкой Х-хромосомы обычно увеличивается с каждым новым поколением, это явление получило название парадокса Шермана.
Патогенез[править | править код]
В начале 1990-х годов было осуществлено секвенирование гена синдрома Мартина — Белл. Полученные результаты показали, что в основе клинических проявлений и цитогенетически выявляемой ломкости X-хромосомы при этом заболевании лежит многократное увеличение числа тринуклеотидных повторов ЦГГ. Оказалось, что у здоровых индивидов число этих повторов в X-хромосоме колеблется от 6 до 54, а увеличение этого числа свыше 200 повторов приводит к феномену ломкой X-хромосомы и клиническому проявлению заболевания. Предмутационное состояние — когда повторов ЦГГ от 55 до 200: заболевание у таких людей в типичной форме не проявляется, но высока вероятность того, что оно проявится у их потомков.
Экспансия тринуклеотидных повторов происходит во время гаметогенеза. Переход от состояния предмутации к полной мутации возможен только при передаче гена от матери, то есть «утяжеление» аллеля происходит во время овогенеза.
Клиническая картина[править | править код]
Мальчик с синдромом Мартина — Белл
Мальчики рождаются с большой массой тела — от 3,5 до 4 кг. Первым признаком, который заставляет заподозрить заболевание, является макроорхизм (увеличение размеров яичек) при отсутствии эндокринной патологии. Также есть определённые фенотипические признаки: большая голова с высоким и широким лбом, длинное лицо с увеличенным подбородком, несколько уплощённая средняя часть лица, тупой, слегка клювовидно загнутый кончик носа. Уши большие, иногда оттопыренные, низко расположенные. Кисти и стопы широкие, дистальные фаланги пальцев также широкие, суставы имеют повышенную подвижность. Кожа нередко гиперэластична. Часто встречаются светлоокрашенные радужные оболочки, светлые волосы. Не обязательно встречаются все признаки — могут быть один или несколько.
Неврологическая симптоматика неспецифична, определяется как и у всех детей с умственной отсталостью. Наблюдается некоторая мышечная гипотония, дискоординация движений. Также могут быть глазодвигательные, пирамидные и экстрапирамидные нарушения.
Главным симптомом синдрома является интеллектуальное недоразвитие и своеобразная речь. Такие больные говорят быстро, сбивчиво, имеются выраженные эхолалии и персеверации (бормочущая речь). Степень умственной отсталости при синдроме Мартина — Белл колеблется между средней и легкой умственной отсталостью[7].
Также могут быть нарушения поведения в виде агрессивности, двигательной расторможенности. В качестве одной из частых психопатологических особенностей отмечена симптоматика, напоминающая аутистическую: стереотипии, эхолалия, мутизм, самоповреждения, трудно устанавливаемый зрительный контакт и непереносимость прикосновений[7]. Однако в отличие от аутистов, эти дети стремятся к общению[7]. Встречаются также подпрыгивания, похлопывания руками, повороты вокруг своей оси, встряхивание кистями, «манежный» бег, разнообразные гримасы, монотонное хныканье.
Кроме вышеописанного у таких детей могут быть признаки раннего детского аутизма.
Диагностика[править | править код]
Характерными особенностями синдрома являются удлинённое лицо, большие или выступающие уши и низкий мышечный тонус.
Синдром хрупкой Х-хромосомы диагностируется путём определения количества ЦГГ-повторов и их статуса метилирования с помощью эндонуклеазной рестрикции и саузерн-блоттинга.
Это заболевание относится к болезням экспансии (экспансия — резкое увеличение числа копий повторяющихся участков молекулы ДНК (повторы) у индивидов в последующих поколениях родословной). Феномен экспансии числа тринуклеотидных повторов (ЦГГ) был впервые обнаружен как раз при молекулярно-генетическом исследовании этого синдрома.
Ранее диагноз синдрома Мартина-Белл основывался на данных клинико-генеалогического анализа и результатах цитогенетического исследования клеток больного, выращенных на специальной среде с дефицитом фолиевой кислоты. В случае обнаружения поломок X-хромосомы в локусе Xq27.3 диагноз синдрома не вызывает сомнений.
Лечение[править | править код]
Лечения для синдрома ломкой Х-хромосомы не существует, однако есть надежда, что дальнейшие исследования причин заболевания предоставят новые возможности терапии. В настоящее время симптомы можно облегчить с помощью когнитивно-поведенческой терапии, специфического обучения, медикаментов и, при необходимости, лечения физических аномалий. Лица, имеющие случаи синдрома ломкой Х-хромосомы в семье, должны получить генетическое консультирование при планировании беременности[8].
Поскольку в эксперименте обнаружение ломкости удалось обнаружить в среде, бедной фолатами, было предложено лечить таких детей фолиевой кислотой.
Эффект от лечения у детей выражен больше, чем у взрослых: пропадает агрессия, повышается внимание, улучшается моторика и речь.
Также пробуют лечить таких больных психостимуляторами.
Примечания[править | править код]
- ↑ Disease Ontology release 2019-05-13 — 2019-05-13 — 2019.
- ↑ Sherman, S. Epidemiology // Fragile X Syndrome, Diagnosis Treatment and Research (англ.) / Hagerman, R. J.; Hagerman, P. J.. — 3rd. — Baltimore: Johns Hopkins University Press (англ.)русск., 2002. — ISBN 0801868432.
- ↑ 1 2 3 J. Purdon Martin, Julia Bell. A pedigree of mental defect showing sex-linkage (англ.) // Journal of Neurology, Neurosurgery, and Psychiatry (англ.)русск. : journal. — 1943. — Vol. 6, no. 3—4. — P. 154—157. — doi:10.1136/jnnp.6.3-4.154. — PMID 21611430.
- ↑ Nolin S.L., Brown W.T., Glicksman A., et al. Expansion of the fragile X CGG repeat in females with premutation or intermediate alleles (англ.) // Am. J. Hum. Genet. (англ.)русск. : journal. — 2003. — Vol. 72, no. 2. — P. 454—464. — doi:10.1086/367713. — PMID 12529854.
- ↑ Bassell G.J., Warren S.T. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function (англ.) // Neuron (англ.)русск. : journal. — Cell Press (англ.)русск., 2008. — Vol. 60, no. 2. — P. 201—214. — doi:10.1016/j.neuron.2008.10.004. — PMID 18957214.
- ↑ Garber K.B., Visootsak J., Warren S.T. Fragile X syndrome (англ.) // Eur J Hum Genet (англ.)русск. : journal. — 2008. — Vol. 16, no. 6. — P. 666. — doi:10.1038/ejhg.2008.61. — PMID 18398441.
- ↑ 1 2 3 Н. Н. Иванец, Ю. Г. Тюльпин, В. В. Чирко, М. А. Кинкулькина. Психиатрия и наркология: учебник. — М.: ГЭОТАР-Медиа, 2006. — С. 596. — 832 с. — ISBN 5-9704-0197-8.
- ↑ Hagerman R.J., Berry-Kravis E., Kaufmann WE et al. Advances in the treatment of fragile X syndrome (англ.) // Pediatrics (англ.)русск.. — American Academy of Pediatrics (англ.)русск., 2009. — Vol. 123, no. 1. — P. 378—390. — doi:10.1542/peds.2008-0317. — PMID 19117905.
Ссылки[править | править код]
- Американские ученые вылечили умственную отсталость у мышей. Medportal.ru
- Синдром ломкой X-хромосомы. Пер. с англ. Н. Д. Фирсовой (2018)
Источник